August, 14, 2023
The ubiquitin-like modifier FAT10 covalently modifies HUWE1 and strengthens the interaction of AMBRA1 and HUWE1.
Stefanie Müller, Johanna Bialas, Stella Ryu, Nicola Catone, and Annette Aichem
PLoS One
14 August 2023, published online
In this study the interaction of the ubiquitin-like modifier FAT10 with the HECT-type ubiquitin E3 ligase HUWE1 and its impact on the actions of the autophagy-related protein AMBRA1 is revealed by scientists of CRC969 projects C01 and C09.
Abstract
The ubiquitin-like modifier FAT10 is highly upregulated under inflammatory conditions and targets its conjugation substrates to the degradation by the 26S proteasome. This process termed FAT10ylation is mediated by an enzymatic cascade and includes the E1 activating enzyme ubiquitin-like modifier activating enzyme 6 (UBA6), the E2 conjugating enzyme UBA6-specific E2 enzyme 1 (USE1) and E3 ligases, such as Parkin. In this study, the function of the HECT-type ubiquitin E3 ligase HUWE1 was investigated as a putative E3 ligase and/or conjugation substrate of FAT10. Our data provide strong evidence that HUWE1 is FAT10ylated in a UBA6 and FAT10 diglycine-dependent manner in vitro and in cellulo and that the HUWE1-FAT10 conjugate is targeted to proteasomal degradation. Since the mutation of all relevant cysteine residues within the HUWE1 HECT domain did not abolish FAT10 conjugation, a role of HUWE1 as E3 ligase for FAT10ylation is rather unlikely. Moreover, we have identified the autophagy-related protein AMBRA1 as a new FAT10 interaction partner. We show that the HUWE1-FAT10 conjugate formation is diminished in presence of AMBRA1, while the interaction between AMBRA1 and HUWE1 is strengthened in presence of FAT10. This implies a putative interplay of all three proteins in cellular processes such as mitophagy.
August, 01, 2023
Specifying conformational heterogeneity of multi-domain proteins at atomic resolution.
Tobias Schneider, Kevin Sawade, Frederic Berner, Christine Peter, and Michael Kovermann
Structure
01 August 2023, published online
Researchers from CRC969 project B09 report on their findings about their MD simulations of the conformational state of diubiquitin molecules which display conformational heterogeneity.
Abstract
The conformational landscape of multi-domain proteins is inherently linked to their specific functions. This also holds for polyubiquitin chains that are assembled by two or more ubiquitin domains connected by a flexible linker thus showing a large interdomain mobility. However, molecular recognition and signal transduction are associated with particular conformational substates that are populated in solution. Here, we apply high-resolution NMR spectroscopy in combination with dual-scale MD simulations to explore the conformational space of K6-, K29-, and K33-linked diubiquitin molecules. The conformational ensembles are evaluated utilizing a paramagnetic cosolute reporting on solvent exposure plus a set of complementary NMR parameters. This approach unravels a conformational heterogeneity of diubiquitins and explains the diversity of structural models that have been determined for K6-, K29-, and K33-linked diubiquitins in free and ligand-bound states so far. We propose a general application of the approach developed here to demystify multi-domain proteins occurring in nature.
June, 23, 2023
NAC controls cotranslational N-terminal methionine excision in eukaryotes.
Martin Gamerdinger, Min Jia,
Science
23 June 2023, published online
The long standing riddle as to how proteins are processed during translation by N-terminal methionine excision has been solved by CRC969 researchers from project A07 in collaboration with scientists from the ETH in Zurich.
Abstract
N-terminal methionine excision from newly synthesized proteins, catalyzed cotranslationally by methionine aminopeptidases (METAPs), is an essential and universally conserved process that plays a key role in cell homeostasis and protein biogenesis. However, how METAPs interact with ribosomes and how their cleavage specificity is ensured is unknown. We discovered that in eukaryotes the nascent polypeptide-associated complex (NAC) controls ribosome binding of METAP1. NAC recruits METAP1 using a long, flexible tail and provides a platform for the formation of an active methionine excision complex at the ribosomal tunnel exit. This mode of interaction ensures the efficient excision of methionine from cytosolic proteins, whereas proteins targeted to the endoplasmic reticulum are spared. Our results suggest a broader mechanism for how access of protein biogenesis factors to translating ribosomes is controlled.
See also the press release of the University of Konstanz!
May, 22, 2023
Calcium-induced compaction and clustering of vesicles tracked with molecular resolution.
Oliva Saldanha, Laura Schiller, and Karin Hauser
Biophysical Journal
22 May 2023, published online
By using attenuated total reflection Fourier-transform infrared (ATR-FTIR) spectroscopy scientists from CRC969 project A02 reveal the influence of Ca2+ on the behaviour of lipid versicles.
Abstract
Theory and simulations predict the complex nature of calcium interaction with the lipid membrane. By maintaining the calcium concentrations at physiological conditions, herein we demonstrate experimentally the effect of Ca2+ in a minimalistic cell-like model. For this purpose, giant unilamellar vesicles (GUVs) with a neutral lipid DOPC are generated and the ion-lipid interaction is observed with attenuated total reflection Fourier-transform infrared (ATR-FTIR) spectroscopy providing molecular resolution. Firstly, Ca2+ encapsulated within the vesicle binds to the phosphate head groups of the inner-leaflets and triggers vesicle compaction. This is tracked by changes in vibrational modes of the lipid groups. As the calcium concentration within the GUV increases, IR intensities change indicating vesicle dehydration and lateral compression of the membrane. Secondly, by inducing a calcium gradient across the membrane up to a ratio of 1:20, interaction between several vesicles occur as Ca2+ can bind to the outer-leaflets leading to vesicle clustering. It is observed that larger calcium gradients induce stronger interactions. These findings with an exemplary biomimetic model reveal that divalent calcium ions not only cause local changes to the lipid packing, but also have macroscopic implications to initiate vesicle-vesicle interaction.
May, 15, 2023
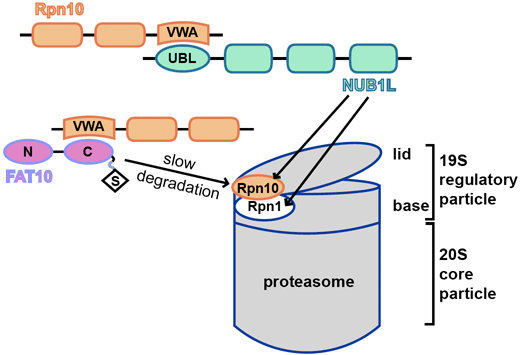
FAT10 and NUB1L cooperate to activate the 26S proteasome.
Life Science Alliance
15 May 2023, published online
CRC969 scientists from projects C01 and C09 report on their findings about the activation mechanism of the 26S proteasome by cooperation of the ubiquitin-like modifier FAT10 and its interaction partner NUB1L. This publication is dedicated to the memory of Marcus Groettrup who passed away in 2022.
Abstract
The interaction of the 19S regulatory particle of the 26S proteasome with ubiquitylated proteins leads to gate opening of the 20S core particle and increases its proteolytic activity by binding of the ubiquitin chain to the inhibitory deubiquitylation enzyme USP14 on the 19S regulatory subunit RPN1. Covalent modification of proteins with the cytokine inducible ubiquitin-like modifier FAT10 is an alternative signal for proteasomal degradation. Here, we report that FAT10 and its interaction partner NUB1L facilitate the gate opening of the 20S proteasome in an ubiquitin- and USP14-independent manner. We also show that FAT10 is capable to activate all peptidolytic activities of the 26S proteasome, however only together with NUB1L, by binding to the UBA domains of NUB1L and thereby interfering with NUB1L dimerization. The binding of FAT10 to NUB1L leads to an increased affinity of NUB1L for the subunit RPN1. In conclusion, the herein described cooperation of FAT10 and NUB1L is a substrate-induced mechanism to activate the 26S proteasome.
April, 12, 2023
Fast conformational clustering of extensive molecular dynamics simulation data.
Simon Hunkler, Kay Diederichs, Oleksandra Kukharenko, and Christine Peter
Journal of Chemical Physics
12 April 2023, published
CRC scientists from project B09 report on their development of a data processing workflow to analyze molecular dynamics simulation data that was specifically designed to find conformational clusters in long molecular simulation data.
Abstract
We present an unsupervised data processing workflow that is specifically designed to obtain a fast conformational clustering of long molecular dynamics simulation trajectories. In this approach, we combine two dimensionality reduction algorithms (cc_analysis and encodermap) with a density-based spatial clustering algorithm (hierarchical density-based spatial clustering of applications with noise). The proposed scheme benefits from the strengths of the three algorithms while avoiding most of the drawbacks of the individual methods. Here, the cc_analysis algorithm is applied for the first time to molecular simulation data. The encodermap algorithm complements cc_analysis by providing an efficient way to process and assign large amounts of data to clusters. The main goal of the procedure is to maximize the number of assigned frames of a given trajectory while keeping a clear conformational identity of the clusters that are found. In practice, we achieve this by using an iterative clustering approach and a tunable root-mean-square-deviation-based criterion in the final cluster assignment. This allows us to find clusters of different densities and different degrees of structural identity. With the help of four protein systems, we illustrate the capability and performance of this clustering workflow: wild-type and thermostable mutant of the Trp-cage protein (TC5b and TC10b), NTL9, and Protein B. Each of these test systems poses their individual challenges to the scheme, which, in total, give a nice overview of the advantages and potential difficulties that can arise when using the proposed method.
Abstract
Ubiquitin chains are flexible multidomain proteins that have important biological functions in cellular signalling. Computational studies with all-atom molecular dynamics simulations of the conformational spaces of polyubiquitins can be challenging due to the system size and a multitude of long-lived meta-stable states. Coarse graining is an efficient approach to overcome this problem-at the cost of losing high-resolution details. Recently, we proposed the back-mapping based sampling (BMBS) approach that reintroduces atomistic information into a given coarse grained (CG) sampling based on a two-dimensional (2D) projection of the conformational landscape, produces an atomistic ensemble and allows to systematically compare the ensembles at the two levels of resolution. Here, we apply BMBS to K48-linked tri-ubiquitin, showing its applicability to larger systems than those it was originally introduced on and demonstrating that the algorithm scales very well with system size. In an extension of the original BMBS we test three different seeding strategies, i.e. different approaches from where in the CG landscape atomistic trajectories are initiated. Furthermore, we apply a recently introduced conformational clustering algorithm to the back-mapped atomistic ensemble. Thus, we obtain insight into the structural composition of the 2D landscape and illustrate that the dimensionality reduction algorithm separates different conformational characteristics very well into different regions of the map. This cluster analysis allows us to show how atomistic trajectories sample conformational states, move through the projection space and in sum converge to an atomistic conformational landscape that slightly differs from the original CG map, indicating a correction of flaws in the CG template.
CRC project B05 develops tools to analyze carbohydrates in living cells by metabolic glycoengineering. In this publication the authors describe the technique they developed to visualize cell-specific sialylation in different classes of membrane constiutents in dopaminergic neurons.
Abstract
Metabolic glycoengineering (MGE) has been developed to visualize carbohydrates on live cells. The method allows the fluorescent labeling of sialic acid (Sia) sugar residues on neuronal plasma membranes. For instance, the efficiency of glycosylation along neurite membranes has been characterized as cell health measure in neurotoxicology. Using human dopaminergic neurons as model system, we asked here, whether it was possible to separately label diverse classes of biomolecules and to visualize them selectively on cells. Several approaches suggest that a large proportion of Sia rather incorporated in non-protein components of cell membranes than into glycoproteins. We made use here of deoxymannojirimycin (dMM), a non-toxic inhibitor of protein glycosylation, and of N-butyl-deoxynojirimycin (NBdNM) a well-tolerated inhibitor of lipid glycosylation, to develop a method of differential labeling of sialylated membrane lipids (lipid-Sia) or sialylated N-glycosylated proteins (protein-Sia) on live neurons. The time resolution at which Sia modification of lipids/proteins was observable was in the range of few hours. The approach was then extended to several other cell types. Using this technique of target-specific MGE, we found that in dopaminergic or sensory neurons >60% of Sia is lipid bound, and thus polysialic acid-neural cell adhesion molecule (PSA-NCAM) cannot be considered the major sialylated membrane component. Different from neurons, most Sia was bound to protein in HepG2 hepatoma cells or in neural crest cells. Thus, our method allows visualization of cell-specific sialylation processes for separate classes of membrane constituents.
In the new publication by scientists from CRC project A02 they decipher the different states of Polyglutamine during aggregation using time-resolved temperature-jump (T-jump) infrared (IR) spectroscopy.
Abstract
Polyglutamine (polyQ) diseases are caused by misfolding and aggregation of expanded polyQ tracts in the affected protein. PolyQ fibrils have been studied in detail; however, less is known about oligomeric precursor states. By a combination of time-resolved temperature-jump (T-jump) infrared (IR) spectroscopy and an appropriately tailored polyQ model peptide, we succeeded in disentangling conformational dynamics in the heterogeneous ensemble of states evolving during aggregation. Individual structural elements could be differentiated by IR-specific signatures, i.e., hairpin monomers, β-structured oligomers, and disordered structure. Submillisecond dynamics were observed for early oligomeric states in contrast to the slow dynamics of fibril growth. We propose that a high structural flexibility of oligomers is required to initiate fibril formation, but not after a fibrillar structure has consolidated and the fibril just grows. Our study reveals that structural flexibility changes at different stages in the aggregation process, from fibril initiation to fibril growth.
April, 15, 2022
Lockdown, a selective small-molecule inhibitor of the integrin phosphatase PPM1F, blocks cancer cell invasion.
Tanja M Grimm, Marleen Herbinger, Lena Krüger, Silke Müller, Thomas U Mayer, and Christof R Hauck
Cell Chem Biol
19 April 2022, online
Scientists from CRC969 project B06 report on their identification of a small-molecule inhibitor of the phosphatase PPM1F which controls cell adhesion and which when upregulated is involved in cancer biology.
Abstract
Phosphatase PPM1F is a regulator of cell adhesion by fine-tuning integrin activity and actin cytoskeleton structures. Elevated expression of this enzyme in human tumors is associated with high invasiveness, enhanced metastasis, and poor prognosis. Thus, PPM1F is a target for pharmacological intervention, yet inhibitors of this enzyme are lacking. Here, we use high-throughput screening to identify Lockdown, a reversible and non-competitive PPM1F inhibitor. Lockdown is selective for PPM1F, because this compound does not inhibit other protein phosphatases in vitro and does not induce additional phenotypes in PPM1F knockout cells. Importantly, Lockdown-treated glioblastoma cells fully re-capitulate the phenotype of PPM1F-deficient cells as assessed by increased phosphorylation of PPM1F substrates and corruption of integrin-dependent cellular processes. Ester modification yields LockdownPro with increased membrane permeability and prodrug-like properties. LockdownPro suppresses tissue invasion by PPM1F-overexpressing human cancer cells, validating PPM1F as a therapeutic target and providing an access point to control tumor cell dissemination.
March, 18, 2022
Generation and characterization of site-specifically mono-ubiquitylated p53.
Alexandra Julier, Vanessa Radtke, Andreas Marx, and Martin Scheffner
ChemBiochem
18 March 2022, published
Researchers from CRC969 project B03 analyzed the effect of mono-ubiquitylation of the tumor suppressor p53 at specific positions on its properties.
Abstract
The tumor suppressor p53 is regulated by various posttranslational modifications including different types of ubiquitylation, which exert distinct effects on p53. While modification by ubiquitin chains targets p53 for degradation, attachment of single ubiquitin moieties (mono-ubiquitylation) affects the intracellular location of p53 and/or its interaction with chromatin. However, how this is achieved at the molecular level remains largely unknown. Similarly, since p53 can be ubiquitylated at different lysine residues, it remains unclear if the eventual effect depends on the position of the lysine modified. Here, we combined genetic code expansion with oxime ligation to generate p53 site-specifically mono-ubiquitylated at position 120. We found that mono-ubiquitylation at this position neither interferes with p53 ubiquitylation by the E3 ligases HDM2 and E6AP in complex with the viral E6 oncoprotein nor affects p53 binding to a cognate DNA sequence. Thus, ubiquitylation per se does not affect physiologically relevant properties of p53.
Mechanism of signal sequence handover from NAC to SRP on ribosomes during ER-protein targeting.
Ahmad Jomaa, Martin Gamerdinger, Hao-Hsuan Hsieh, Annalena Wallisch,Viswanathan Chandrasekaran, Zeynel Ulusoy, Alain Scaiola, Ramanujan S. Hegde, Shu-ou Shan,Nenad Ban, and Elke Deuerling
Science
25 February 2022, published
Researchers from CRC969 project A07 in collaboration with scientists from the ETH Zurich, the MRC Laboratory in Cambridge and the Caltech in Pasadena solve the long-standig riddle on how NAC controls the sorting of nascent polypeptides to their correct destination inside the cell.
Abstract
The nascent polypeptide-associated complex (NAC) interacts with newly synthesized proteins at the ribosomal tunnel exit and competes with the signal recognition particle (SRP) to prevent mistargeting of cytosolic and mitochondrial polypeptides to the endoplasmic reticulum (ER). How NAC antagonizes SRP and how this is overcome by ER targeting signals are unknown. Here, we found that NAC uses two domains with opposing effects to control SRP access. The core globular domain prevented SRP from binding to signal-less ribosomes, whereas a flexibly attached domain transiently captured SRP to permit scanning of nascent chains. The emergence of an ER-targeting signal destabilized NAC’s globular domain and facilitated SRP access to the nascent chain. These findings elucidate how NAC hands over the signal sequence to SRP and imparts specificity of protein localization.
see also the Press release by the University of Konstanz
February, 18, 2022
PolyQ aggregation studied by model peptides with intrinsic tryptophan fluorophores.
Ho-Wah Siu, Paul Stritt, Heng Zhao, and Karin Hauser
Biophysical Chemistry, published online
PolyQ-rich hairpin peptides were designed by scientists from CRC969 project A02 to analyze the effect of the number of glutamines present on the structural stability and aggregation of polyQ repeats.
Abstract
Polyglutamine (polyQ) model peptides are ideally suited to analyze the involvement of glutamines in the disease-related aggregation onset. Here we use a template-assisted design of polyQ-rich hairpin peptides (Trpzip-Qn) to monitor structural stability with fluorescence spectroscopy. The hairpin model imitates the monomeric motif of a polyQ fibril and is stabilized by hydrophobic interactions of two cross-strand pairs of tryptophans (Trps) which are used as fluorophores to report on structural changes. The Trps also frame the polyQ repeats located on each hairpin strand with a different number of glutamines (Qn). Single-stranded sequences mimic the unfolded state and were used as references to differentiate the intrinsic fluorescence signal from the spectral effect caused by structural changes. Temperature-induced hairpin unfolding was monitored by the spectral shift of the Trp fluorescence signal and transition temperatures were determined. The magnitude of the spectral shift indicates the degree of structural disorder. We observed that a longer polyQ repeat is more disordered and weakens the cross-strand Trp-Trp interactions resulting in a decrease of the spectral shift. Aggregation to a fibrillar and more ordered structure shows an increase of the spectral shift. In addition, a band at 280 nm occurs in the spectrum which clearly correlates with the turbidity of the sample and is attributed to scattering of larger aggregated structures. Our study reveals that the number of glutamines, pH and temperature affect structural stability and aggregation of polyQ repeats.
Cell Rep
16 March 2021, published online
CRC969 scientists from projects A06 and C01 teamed up to analyze the role of the ubiquitin-like modifier FAT10 in the control of the E3 ligase Parkin.
Abstract
Parkin is an E3 ubiquitin ligase belonging to the RING-between-RING family. Mutations in the Parkin-encoding gene PARK2 are associated with familial Parkinson’s disease. Here, we investigate the interplay between Parkin and the inflammatory cytokine-induced ubiquitin-like modifier FAT10. FAT10 targets hundreds of proteins for degradation by the 26S proteasome. We show that FAT10 gets conjugated to Parkin and mediates its degradation in a proteasome-dependent manner. Parkin binds to the E2 enzyme of FAT10 (USE1), auto-FAT10ylates itself, and facilitates FAT10ylation of the Parkin substrate Mitofusin2 in vitro and in cells, thus identifying Parkin as a FAT10 E3 ligase. On mitochondrial depolarization, FAT10ylation of Parkin inhibits its activation and ubiquitin-ligase activity causing impairment of mitophagy progression and aggravation of rotenone-mediated death of dopaminergic neuronal cells. In conclusion, FAT10ylation inhibits Parkin and mitophagy rendering FAT10 a likely inflammation-induced exacerbating factor and potential drug target for Parkinson’s disease.
February, 26, 2021
Snapshots of native pre-50S ribosomes reveal a biogenesis factor network and evolutionary specialization.
Mol Cell
26 February 2021, published online
In a collaborative effort scientists from CRC969 project A01 together with researchers from different research institutes in Berlin characterized a network of biogenesis factors involved in the maturation of 50S ribosomes.
Abstract
Ribosome biogenesis is a fundamental multi-step cellular process that culminates in the formation of ribosomal subunits, whose production and modification are regulated by numerous biogenesis factors. In this study, we analyze physiologic prokaryotic ribosome biogenesis by isolating bona fide pre-50S subunits from an Escherichia coli strain with the biogenesis factor ObgE, affinity tagged at its native gene locus. Our integrative structural approach reveals a network of interacting biogenesis factors consisting of YjgA, RluD, RsfS, and ObgE on the immature pre-50S subunit. In addition, our study provides mechanistic insight into how the GTPase ObgE, in concert with other biogenesis factors, facilitates the maturation of the 50S functional core and reveals both conserved and divergent evolutionary features of ribosome biogenesis between prokaryotes and eukaryotes.
December, 21, 2020
A ligand selection strategy identifies chemical probes targeting the proteases of SARS-CoV-2.
Dávid
Angew Chem Int Ed
21 December 2020, published online
In a collaborative effort researchers from CRC969 projects B09 and C06 developed activity-based probes targeting two cysteine proteases of SARS-CoV-2.
Abstract
Activity-based probes are valuable tools for chemical biology. However, finding probes that specifically target the active site of an enzyme remains a challenging task. Here we present a ligand selection strategy that allows to rapidly tailor electrophilic probes to a target of choice and showcase its application for the two cysteine proteases of SARS-CoV-2 as proof of concept. The resulting probes were specific for the active site labelling of 3CL pro and PL pro with sufficient selectivity in a live cell model as well as in the background of a native human proteome. Exploiting the probes as tools for competitive profiling of a natural product library identified salvianolic acid derivatives as promising 3CL pro inhibitors. We anticipate that our ligand selection strategy will be useful to rapidly develop customized probes and discover inhibitors for a wide range of target proteins also beyond corona virus proteases.
October, 28, 2020
Template-assisted design of monomeric polyQ models to unravel the unique role of glutamine side chains in disease-related aggregation.
Hoh-Wah Siu, Benjamin Heck, Michael Kovermann and Karin Hauser
Chem Sci
28 October 2020, published online
CRC scientists from Project A02 and Project B09 collaborated to elucidate the role of glutamine side chains in the aggregation of monomeric polyQ model sequences.
Abstract
Expanded polyglutamine (polyQ) sequences cause numerous neurodegenerative diseases which are accompanied by the formation of polyQ fibrils. The unique role of glutamines in the aggregation onset is undoubtedly accepted and a lot structural data of the fibrils have been acquired, however side-chain specific structural dynamics inducing oligomerization are not well understood yet. To analyze spectroscopically the nucleation process, we designed various template-assisted glutamine-rich β–hairpin monomers mimicking the structural motif of a polyQ fibril. In a top-down strategy, we use a template which forms a well-defined stable hairpin in solution, insert polyQ-rich sequences into each strand and monitor the effects of individual glutamines by NMR, CD and IR spectroscopic approaches. The design was further advanced by alternating glutamines with other amino acids (T, W, E, K), thereby enhancing the solubility and increasing the number of cross-strand interacting glutamine side chains. Our spectroscopic studies reveal a decreasing hairpin stability with increased glutamine content and demonstrate the enormous impact of only a few glutamines – far below the disease threshold – to destabilize structure. Furthermore, we could access sub-ms conformational dynamics of monomeric polyQ-rich peptides by laser-excited temperature-jump IR spectroscopy. Both, the increased number of interacting glutamines and higher concentrations are key parameters to induce oligomerization. Concentration-dependent time-resolved IR measurements indicate an additional slower kinetic phase upon oligomer formation. The here presented peptide models enable spectroscopic molecular analyses to distinguish between monomer and oligomer dynamics in the early steps of polyQ fibril formation and in a side-chain specific manner.
August, 14, 2020
The ubiquitin-like modifier FAT10 inhibits retinal PDE6 activity and mediates its proteasomal degradation.
Annika Böhm, Johanna Bialas, Nicola Catone, Almudena Sacristán-Reviriego, Jacqueline van der Spuy, Marcus Groettrup and Annette Aichem
J Biol Chem
14 August 2020, published online
Researchers from CRC Project C01 in collaboration with scientists from the University College London reveal the control of the retinal cGMP phosphodiesterase PDE6 by the ubiquitin-like modifier FAT10.
Abstract
The retina-specific chaperone AIPL1 is essential for the correct assembly of phosphodiesterase 6 (PDE6), which is a pivotal effector enzyme for phototransduction and vision because it hydrolyzes cGMP. AIPL1 interacts with the cytokine-inducible ubiquitin-like modifier FAT10 that gets covalently conjugated to hundreds of proteins and targets its conjugation substrates for proteasomal degradation, but whether FAT10 affects PDE6 function or turnover is unknown. Here, we show that FAT10 mRNA is expressed in human retina and identify rod PDE6 as a retina-specific substrate of FAT10 conjugation. We found that AIPL1 stabilizes the FAT10 monomer as well as the PDE6-FAT10 conjugate. Additionally, we elucidated the functional consequences of PDE6 FAT10ylation. On the one hand, we demonstrate that FAT10 targets PDE6 for proteasomal degradation by formation of a covalent isopeptide linkage. On the other hand, FAT10 inhibits PDE6 cGMP hydrolyzing activity by non-covalently interacting with the PDE6 GAFa and catalytic domains. Therefore, FAT10 may contribute to loss of PDE6 and, as a consequence, degeneration of retinal cells in eye diseases linked to inflammation and inherited blindness causing mutations in AIPL1.
May, 1, 2020
Real-time monitoring of PARP1-dependent PARylation by ATR-FTIR spectroscopy.
Annika Krüger, Alexander Bürkle, Karin Hauser and Aswin Mangerich
Nat Commun
1 May 2020, published online
Scientists from CRC Project A02 and the biology department of the University of Konstanz report on their findings on PARylation of DNA analyzed in real-time using advanced infrared spectroscopic methods. See also here.
Abstract
Poly-ADP-ribosylation (PARylation) is a fully reversible post-translational modification with key roles in cellular physiology. Due to the multi-domain structure of poly(ADP-ribose) polymerase-1 (PARP1) and the highly dynamic nature of the PARylation reaction, studies on the biochemical mechanism and structural dynamics remain challenging. Here, we report label-free, time-resolved monitoring of PARP1-dependent PARylation using ATR-FTIR spectroscopy. This includes PARP1 activation by binding to DNA strand break models, NAD+ substrate binding, PAR formation, and dissociation of automodified PARP1 from DNA. Analyses of PARP1 activation at different DNA models demonstrate a strong positive correlation of PARylation and PARP1 dissociation, with the strongest effects observed for DNA nicks and 3’ phosphorylated ends. Moreover, by examining dynamic structural changes of PARP1, we reveal changes in the secondary structure of PARP1 induced by NAD+ and PARP inhibitor binding. In summary, this approach enables holistic and dynamic insights into PARP1-dependent PARylation with molecular and temporal resolution.
April, 7, 2020
The length of a ubiquitin chain is a determinant for selective recognition by ubiquitin-binding proteins.
Joachim Lutz, Eva Höllmüller, Martin Scheffner, Andreas Marx and Florian Stengel
Angew Chem Int Ed
7 April 2020, published online
CRC969 scientists from Project A06 and Project B03 discovered that the length of a ubiquitin chain attached to a protein is of crucial importance for its recognition by ubiquitin binding proteins.
Abstract
The attachment of differently linked ubiquitin (Ub) chains of varying length to proteins is a prevalent posttranslational modification in eukaryotic cells. The fate of a modified protein is determined by Ub‐binding proteins (UBPs) that interact with Ub chains in a linkage‐selective manner. Therefore, proteome‐wide interaction studies using differently linked Ub chains have become an increased focus of research activities. However, the impact and functional consequences of chain length on the binding selectivity of UBPs remain mostly elusive, due to a lack of available tools and sufficient amounts of pure, length‐defined Ub chains. Here, we generated linkage‐ and length‐defined Ub chains using click‐chemistry and Gel Eluted Liquid Fraction Entrapment Elelectrophoresis (GELFrEE) fractionation and employed such defined polymers in affinity‐based enrichment assays to identify length‐ and linkage‐selective interactors on a proteome‐wide scale. For the first time, this revealed that the length of a Ub chain has generally a major impact on its ability to be selectively recognized by UBPs.
February, 18, 2020
Proteome-wide structural probing of low-abundant protein interactions by cross-linking mass spectrometry.
Julius Fürsch, Kai-Michael Kammer, Stefan G. Kreft, Martin Beck and Florian Stengel
Anal Chem
18 February 2020, published online
Researchers from Project A06 together with scientists from the EMBL Heidelberg report about their achievements in probing of of low-abundant proteins by cross-linking mass spectrometry.
Abstract
Proteome-wide cross-linking studies have spurred great interest as they facilitate structural probing of protein interactions in living cells and organisms. However, current studies have a bias for high-abundant proteins. In this study we demonstrate both experimentally and by a kinetic model that this bias is also caused by the propensity of cross-links to preferentially form on high abundant proteins and not by the inability to detect cross-links due to limitations in current technology. We further show, by using both an in vitro mimic of a crowded cellular environment and eukaryotic cell lysates, that parameters optimized toward a pseudo first order kinetics model result in a significant increase in the detection of lower-abundant proteins on a proteome-wide scale. Our study therefore explains the cause of a major limitation in current proteome-wide cross-linking studies and demonstrates how to address a larger part of the proteome by cross-linking.
January, 21, 2020
Competitive metabolite profiling of natural products reveals subunit specific inhibitors of the 20S proteasome
Atul Pawar, Michael Basler, Heike Goebel, Gerardo Omar Alvarez Salinas, Marcus Groettrup and Thomas Böttcher
ACS Cent Sci
21 January 2020, published online
In a collaborative effort by CRC scientists from Project C06 and Project C01 inhibitors of the 20S proteasome were indentified.
Abstract
We have developed a syringolin-based chemical probe and explored its utility for the profiling of metabolite extracts as potent inhibitors of the 20S proteasome. Activity-guided fractionation by competitive labeling allowed us to isolate and identify glidobactin A and C as well as luminmycin A from a Burkholderiales strain. The natural products exhibited unique subunit specificities for the proteolytic subunits of human and mouse constitutive and immunoproteasome in the lower nanomolar range. In particular, glidobactin C displayed an unprecedented β2/β5 coinhibition profile with single-digit nanomolar potency in combination with sufficiently high cell permeability. These properties render glidobactin C a promising live cell proteasome inhibitor with potent activity against human breast cancer cell lines and comparably low immunotoxicity.
December, 27, 2019
Conformational and functional characterization of artificially conjugated non-canonical ubiquitin dimers
Tobias Schneider, Andrej Berg, Zeynel Ulusoy, Martin Gamerdinger, Christine Peter and Michael Kovermann
Sci Rep
27 December 2019, published online
By combining NMR spectrocopy with MD simulations researchers from Project A07 and Project B09 characrterized non-canonical ubiquitin dimers.
Abstract
Ubiquitylation is an eminent posttranslational modification referring to the covalent attachment of single ubiquitin molecules or polyubiquitin chains to a target protein dictating the fate of such labeled polypeptide chains. Here, we have biochemically produced artificially Lys11-, and Lys27-, and Lys63-linked ubiquitin dimers based on click-chemistry generating milligram quantities in high purity. We show that the artificial linkage used for the conjugation of two ubiquitin moieties represents a fully reliable surrogate of the natural isopeptide bond by acquiring highly resolved nuclear magnetic resonance (NMR) spectroscopic data including ligand binding studies. Extensive coarse grained and atomistic molecular dynamics (MD) simulations allow to extract structures representing the ensemble of domain-domain conformations used to verify the experimental data. Advantageously, this methodology does not require individual isotopic labeling of both ubiquitin moieties as NMR data have been acquired on the isotopically labeled proximal moiety and complementary MD simulations have been used to fully interpret the experimental data in terms of domain-domain conformation. This combined approach intertwining NMR spectroscopy with MD simulations makes it possible to describe the conformational space non-canonically Lys11-, and Lys27-linked ubiquitin dimers occupy in a solution averaged ensemble by taking atomically resolved information representing all residues in ubiquitin dimers into account.
November, 08, 2019
A Theophylline-Responsive Riboswitch Regulates Expression of Nuclear-Encoded Genes.
Plant Physiol
08 November 2019, published online
CRC 969 scientists from Project A05 and former Project C02 have developed synthetic riboswitches as tools for regulation of nuclear gene expression in plants.
Abstract
Riboswitches are small cis-regulatory RNA elements that regulate gene expression by conformational changes in response to ligand binding. Synthetic riboswitches have been engineered as versatile and innovative tools for gene regulation by external application of their ligand in prokaryotes and eukaryotes. In plants, synthetic riboswitches were used to regulate gene expression in plastids, but the application of synthetic riboswitches for the regulation of nuclear-encoded genes in planta remains to be explored. Here, we characterize the properties of a theophylline-responsive synthetic aptazyme for control of nuclear-encoded transgenes in Arabidopsis (Arabidopsis thaliana). Activation of the aptazyme, inserted in the 3′ UTR of the target gene, resulted in rapid self-cleavage and subsequent decay of the mRNA. This riboswitch allowed reversible, theophylline-dependent down-regulation of the GFP reporter gene in a dose- and time-dependent manner. Insertion of the riboswitch into the ONE HELIX PROTEIN1 gene allowed complementation of ohp1 mutants and induction of the mutant phenotype by theophylline. GFP and ONE HELIX PROTEIN1 transcript levels were downregulated by up to 90%, and GFP protein levels by 95%. These results establish artificial riboswitches as tools for externally controlled gene expression in synthetic biology in plants or functional crop design.
September, 04, 2019
Immunoproteasome inhibition selectively kills human CD14+ monocytes and as a result dampens IL-23 secretion
Michael Basler, Meike Claus, Moritz Klawitter, Heike Goebel and Marcus Groettrup
J Immunol
04 Sept 2019, published online
Scientists from Project C01 unravel the role of immunoproteasome inhibition in IL-23-driven autoimmunity.
Abstract
MECL-1 (β2i), LMP2 (β1i), and LMP7 (β5i) are the proteolytically active subunits of the immunoproteasome (IP), a special type of proteasome mainly expressed in hematopoietic cells. Targeting the IP in autoimmune diseases proved to be therapeutically effective in preclinical mouse models. In endotoxin-stimulated human PBMCs, IP inhibition reduces the secretion of several proinflammatory cytokines, with the suppression of IL-23 being the most prominent. In this study, we investigated why the production of IL-23, a key mediator of inflammation in autoimmunity, is blocked when the IP is inhibited in LPS-stimulated human PBMCs. CD14+ monocytes could be identified as the main producers of IL-23 in LPS-stimulated PBMCs. We found that IP inhibition with the irreversible LMP7/LMP2 inhibitor ONX 0914 induced apoptosis in CD14+ monocytes, whereas CD4+, CD3+, CD19+, and CD56+ cells remained unaffected. A high expression of IPs renders monocytes susceptible to IP inhibition, leading to an accumulation of polyubiquitylated proteins and the induction of the unfolded protein response. Similar to IP inhibition, inducers of the unfolded protein response selectively kill CD14+ monocytes in human PBMCs. The blockage of the translation in CD14+ monocytes protects these cells from ONX 0914–induced cell death, indicating that the IP is required to maintain protein turnover in monocytes. Taken together, our data reveal why IP inhibition is particularly effective in the suppression of IL-23–driven autoimmunity.
July, 31, 2019
Early scanning of nascent polypeptides inside the ribosomal tunnel by NAC
Martin Gamerdinger, Kan Kobayashi, Annalena Wallisch, Stefan G. Kreft, Carolin Sailer, Renate Schlömer, Nadine Sachs, Ahmad Jomaa, Florian Stengel, Nenad Ban and Elke Deuerling
Mol Cell
31 July 2019, published online
Scientists from Project A07 and Project A06 together with colleagues from the ETH Zurich discovered that the N-terminal tail of the β subunit of the nascent polypeptide-associated complex can act as a sensor of substrates directly upon synthesis by the ribosome.
Abstract
Cotranslational processing of newly synthesized proteins is fundamental for correct protein maturation. Protein biogenesis factors are thought to bind nascent polypeptides not before they exit the ribosomal tunnel. Here, we identify a nascent chain recognition mechanism deep inside the ribosomal tunnel by an essential eukaryotic cytosolic chaperone. The nascent polypeptide-associated complex (NAC) inserts the N-terminal tail of its β subunit (N-βNAC) into the ribosomal tunnel to sense substrates directly upon synthesis close to the peptidyl-transferase center. N-βNAC escorts the growing polypeptide to the cytosol and relocates to an alternate binding site on the ribosomal surface. Using C. elegans as an in vivo model, we demonstrate that the tunnel-probing activity of NAC is essential for organismal viability and critical to regulate endoplasmic reticulum (ER) protein transport by controlling ribosome-Sec61 translocon interactions. Thus, eukaryotic protein maturation relies on the early sampling of nascent chains inside the ribosomal tunnel.
June, 10, 2019
Reductive modification of genetically encoded 3-nitrotyrosine sites in alpha synuclein expressed in E. coli
Hanne R. Gerding, Christiaan Karreman, Andreas Daiber, Johannes Delp, Daniel Hammler, Martin Mex, Stefan Schildknecht and Marcel Leist
Redox Biol
10 June 2019, published online
Scientists from Project C05 report on the capability of E. coli cells to rapidly perform a reductive modification on seberal ectopically expressed proteins, amongst them nitrated alpha-synuclein.
Abstract
Tyrosine nitration is a post-translational protein modification relevant to various pathophysiological processes. Chemical nitration procedures have been used to generate and study nitrated proteins, but these methods regularly lead to modifications at other amino acid residues. A novel strategy employs a genetic code modification that allows incorporation of 3-nitrotyrosine (3-NT) during ribosomal protein synthesis to generate a recombinant protein with defined 3-NT-sites, in the absence of other post-translational modifications. This approach was applied to study the generation and stability of the 3-NT moiety in recombinant proteins produced in E.coli. Nitrated alpha-synuclein (ASYN) was selected as exemplary protein, relevant in Parkinson’s disease (PD). A procedure was established to obtain pure tyrosine-modified ASYN in mg amounts. However, a rapid (t1/2 = 0.4 h) reduction of 3-NT to 3-aminotyrosine (3-AT) was observed. When screening for potential mechanisms, we found that 3-NT can be reduced enzymatically to 3-AT, whilst biologically relevant low molecular weight reductants, such as NADPH or GSH, did not affect 3-NT. A genetic screen for E.coli proteins, involved in the observed 3-NT reduction, revealed the contribution of several, possibly redundant pathways. Green fluorescent protein was studied as an alternative model protein. These data confirm 3-NT reduction as a broadly-relevant pathway in E. coli. In conclusion, incorporation of 3-NT as a genetically-encoded non-natural amino acid allows for generation of recombinant proteins with specific nitration sites. The potential reduction of the 3-NT moiety by E.coli, however, requires attention to the design of the purification strategy for obtaining pure nitrated protein.
May, 31, 2019
Immobilization approaches can affect proteindynamics: a surface-enhanced infrared spectroscopic study on lipid–protein interactions
Mohammad A Fallah and Karin Hauser
Biomat Sci
31 May 2019, published online
Researchers from Project A02 demonstrate that lipid induced conformational changes of α-synuclein are hindered when the protein is immobilized. This has to be considered when applying surface immobilization procedures which are commonly used in many analytical applications.
Abstract
The intrinsically disordered Parkinson disease protein α-synuclein (αS) performs conformational changes induced by intermolecular protein–protein as well as by protein-membrane interactions. Aggregation of αS is a hallmark for the disease, however the role of the membrane in the aggregation process still needs to be clarified. We used a surface-enhanced infrared absorption (SEIRA) spectroscopic approach to investigate the effect of lipid interactions on αS conformation. The near-field detection of SEIRA allows to study exclusively structural changes of immobilized αS with the advantage that the supernatant remains undetected and thus does not interfere with the spectral read-out. Self-assembled monolayer (SAMs) of mixed NHS-PEG-SH linker and MT(PEG)4 spacer molecules were utilized to immobilize αS. The linker/spacer composition of the SAM was adjusted to prevent αS–αS interactions. Two different methods were applied for site-specific (C-terminal and N-terminal) αS immobilization. The immobilized protein was then exposed to lipid vesicles and SEIRA difference spectra were recorded to monitor the αS conformation over time. Irrespective of the used immobilization method, αS tethering hindered lipid-induced conformational changes. The spectra also indicate that a fraction of the immobilized αS eventually desorbs from the surface into the supernatant solution. Desorbed αS performs conformational changes and formation of β-structured aggregates is observed upon interaction with either lipid vesicles or supplementary αS. Our study demonstrates that αS aggregates only when the protein is free in solution and that surface immobilization procedures, commonly used in many analytical applications, can change the dynamic behavior of proteins thereby affecting protein structure and function.
May, 22, 2019
Mechanism of completion of peptidyltransferase centre assembly in eukaryotes
eLife
22 May 2019, published online
In a collaborative effort by researchers from across the European Union including the Cambridge Institute for Medical ResearchUniversity of Cambridge, researchers from Project A06, the University of Graz, the Maria Curie-Skłodowska University, MRC Laboratory of Molecular Biology and the University of Leeds the process of completion of the peptidyltransferase center was unraveled.
Abstract
During their final maturation in the cytoplasm, pre-60S ribosomal particles are converted to translation-competent large ribosomal subunits. Here, we present the mechanism of peptidyltransferase centre (PTC) completion that explains how integration of the last ribosomal proteins is coupled to release of the nuclear export adaptor Nmd3. Single-particle cryo-EM reveals that eL40 recruitment stabilises helix 89 to form the uL16 binding site. The loading of uL16 unhooks helix 38 from Nmd3 to adopt its mature conformation. In turn, partial retraction of the L1 stalk is coupled to a conformational switch in Nmd3 that allows the uL16 P-site loop to fully accommodate into the PTC where it competes with Nmd3 for an overlapping binding site (base A2971). Our data reveal how the central functional site of the ribosome is sculpted and suggest how the formation of translation-competent 60S subunits is disrupted in leukaemia-associated ribosomopathies.
May, 21, 2019
Interactions of p53 with poly(ADP-ribose) and DNA induce distinct changes in protein structure as revealed by ATR-FTIR spectroscopy
Nucleic Acids Res
21 May 2019, published online
The technique of ATF-FTIR was used by scientists from Project A02 and former Project B04 to analyze the changes in the secondary structure of the protein p53 by non-covalent interaction with DNA and poly(ADP-ribose).
Abstract
Due to multiple domains and in part intrinsically disordered regions, structural analyses of p53 remain a challenging task, particularly in complex with DNA and other macromolecules. Here, we applied a novel attenuated total reflection Fourier transform infrared (ATR-FTIR) spectroscopic approach to investigate changes in secondary structure of full-length p53 induced by non-covalent interactions with DNA and poly(ADP-ribose) (PAR). To validate our approach, we confirmed a positive regulatory function of p53’s C-terminal domain (CTD) with regard to sequence-specific DNA binding and verified that the CTD mediates p53–PAR interaction. Further, we demonstrate that DNA and PAR interactions result in distinct structural changes of p53, indicating specific binding mechanisms via different domains. A time-dependent analysis of the interplay of DNA and PAR binding to p53 revealed that PAR represents p53’s preferred binding partner, which efficiently controls p53–DNA interaction. Moreover, we provide infrared spectroscopic data on PAR pointing to the absence of regular secondary structural elements. Finally, temperature-induced melting experiments via CD spectroscopy show that DNA binding stabilizes the structure of p53, while PAR binding can shift the irreversible formation of insoluble p53 aggregates to higher temperatures. In conclusion, this study provides detailed insights into the dynamic interplay of p53 binding to DNA and PAR at a formerly inaccessible molecular level.
May, 15, 2019
Protein spin labeling with a photocaged nitroxide using Diels‐Alder chemistry
Anandi Kugele, Bjarne Silkenath, Jakob Langer, Valentin Wittmann and Malte Drescher
ChemBioChem
15 May 2019, published online
Researchers from Project C03 have established a new strategy to spin-label proteins site-directedly by applying inverse-electron-demand Diels-Alder cycloaddition. See also here.
Abstract
EPR spectroscopy of diamagnetic bio‐macromolecules is based on site‐directed spin labeling (SDSL). Here, we present a novel labeling strategy for proteins. We developed and synthesized a nitroxide‐based spin label that can be ligated to proteins by an inverse‐electron‐demand Diels‐Alder (DAinv) cycloaddition to genetically encoded non‐canonical amino acids (ncAA). The nitroxide moiety is shielded by a photoremovable protecting group (PPG) with an attached tetraethylene glycol unit to achieve water solubility. We demonstrate SDSL of two model proteins with the PaNDA (Photoactivatable Nitroxide for DAinv reaction) label. Our strategy features high reaction rates combined with high selectivity, and the possibility to deprotect the nitroxide in E. coli lysate.
April, 19, 2019
Simulating and analysing configurational landscapes of protein–protein contact formation
Andrej Berg and Christine Peter
Interface Focus
19 April 2019, published online
In Project B09 molecular simulation is used to understand the formation and to characterize the configurational ensemble of protein aggregates. Here the researchers report on their characterization of ubiquitin dimers.
Abstract
Interacting proteins can form aggregates and protein–protein interfaces with multiple patterns and different stabilities. Using molecular simulation one would like to understand the formation of these aggregates and which of the observed states are relevant for protein function and recognition. To characterize the complex configurational ensemble of protein aggregates, one needs a quantitative measure for the similarity of structures. We present well-suited descriptors that capture the essential features of non-covalent protein contact formation and domain motion. This set of collective variables is used with a nonlinear multi-dimensional scaling-based dimensionality reduction technique to obtain a low-dimensional representation of the configurational landscape of two ubiquitin proteins from coarse-grained simulations. We show that this two-dimensional representation is a powerful basis to identify meaningful states in the ensemble of aggregated structures and to calculate distributions and free energy landscapes for different sets of simulations. By using a measure to quantitatively compare free energy landscapes we can show how the introduction of a covalent bond between two ubiquitin proteins at different positions alters the configurational states of these dimers.
April, 18, 2019
Double nitroxide labeling by copper-catalyzed azide–alkyne cycloadditions with noncanonical amino acids for Electron Paramagnetic Resonance Spectroscopy
Pia Widder, Frederic Berner, Daniel Summerer and Malte Drescher
ACS Chem Biol
18 April 2019, published online
Scientists from Project C03 have identified different noncanonical amino acids as suitable tools for site-directed spin labeling of proteins to be analyzed by electron paramagnetic resonance spectroscopy.
Abstract
Electron paramagnetic resonance spectroscopy in combination with site-directed spin labeling (SDSL) is an important tool to obtain long-range distance restraints for protein structural research. We here study a variety of azide- and alkyne-bearing noncanonical amino acids (ncAA) in terms of protein single- and double-incorporation efficiency via nonsense suppression, metabolic stability, yields of nitroxide labeling via copper-catalyzed [3 + 2] azide–alkyne cycloadditions (CuAAC), and spectroscopic properties in continuous-wave and double electron–electron resonance measurements. We identify para-ethynyl-l-phenylalanine and para-propargyloxy-l-phenylalanine as suitable ncAA for CuAAC-based SDSL that will complement current SDSL approaches, particularly in cases in which essential cysteines of a target protein prevent the use of sulfhydryl-reactive spin labels.
April, 11, 2019
Dual role of ribosome-binding domain of NAC as a potent suppressor of protein aggregation and aging-related proteinopathies
Koning Shen, Martin Gamerdinger, Rebecca Chan, KarinaGense, Esther M. Martin, Nadine Sachs, Patrick D. Knight, Renate Schlömer, Antonio N. Calabrese, Katie L. Stewart, Lukas Leiendecker, Ankit Baghel, Sheena E. Radford, Judith Frydman and Elke Deuerling
Mol Cell
11 April 2019, published online
Scientists from Project A07 in collaboration with researchers from the University of Leeds and Stanford University report on their finding that the positively charged N-terminus of the βsubunit of the nascent polypeptide-associated complex represents a major chaperone entity of NAC.
Abstract
The nascent polypeptide-associated complex (NAC) is a conserved ribosome-associated protein biogenesis factor. Whether NAC exerts chaperone activity and whether this function is restricted to de novo protein synthesis is unknown. Here, we demonstrate that NAC directly exerts chaperone activity toward structurally diverse model substrates including polyglutamine (PolyQ) proteins, firefly luciferase, and Aβ40. Strikingly, we identified the positively charged ribosome-binding domain in the N terminus of the βNAC subunit (N-βNAC) as a major chaperone entity of NAC. N-βNAC by itself suppressed aggregation of PolyQ-expanded proteins in vitro, and the positive charge of this domain was critical for this activity. Moreover, we found that NAC also exerts a ribosome-independent chaperone function in vivo. Consistently, we found that a substantial fraction of NAC is non-ribosomal bound in higher eukaryotes. In sum, NAC is a potent suppressor of aggregation and proteotoxicity of mutant PolyQ-expanded proteins associated with human diseases like Huntington’s disease and spinocerebellar ataxias.
April, 5, 2019
Direct imaging of protein‐specific methylation in mammalian cells
Franziska Doll, Raphael R Steimbach and Andreas Zumbusch
ChemBioChem
5 April 2019, published online
Using advanced flourescence imaging techniques, researchers from Project B08 report on their results on imaging of methylation in living cells.
Abstract
Abundant post‐translational modification through methylation alters the function, stability, and/or localization of a protein. Malfunctions in post‐translational modification are associated with severe diseases. To unravel protein methylation sites and their biological functions, chemical methylation reporters have been developed. However, until now, their usage was limited to cell lysates. Herein, we present the first generally applicable approach for imaging methylation of individual proteins in human cells, which is based on a combination of chemical reporter strategies, bioorthogonal ligation reactions, and FRET detected by means of fluorescence lifetime imaging microscopy. Through this approach, methylation of histone 4 and the non‐histone proteins tumor suppressor p53, kinase Akt1, and transcription factor Foxo1 in two human cell lines has been successfully imaged. To further demonstrate its potential, the localization‐dependent methylation state of Foxo1 in the cellular context has been visualized.
March, 4, 2019
Cyclopropene derivatives of aminosugars for metabolic glycoengineering
Jessica Hassenrück and Valentin Wittmann
Beilstein J Org Chem
4 March 2019, published online
Project B05 studies metabolic glycoengineering. Now researchers report on their analysis of different derivatives of cyclopropene-modified hexosamine.
Abstract
Cyclopropenes have been proven valuable chemical reporter groups for metabolic glycoengineering (MGE). They readily react with tetrazines in an inverse electron-demand Diels–Alder (DAinv) reaction, a prime example of a bioorthogonal ligation reaction, allowing their visualization in biological systems. Here, we present a comparative study of six cyclopropene-modified hexosamine derivatives and their suitability for MGE. Three mannosamine derivatives in which the cyclopropene moiety is attached to the sugar by either an amide or a carbamate linkage and that differ by the presence or absence of a stabilizing methyl group at the double bond have been examined. We determined their DAinv reaction kinetics and their labeling intensities after metabolic incorporation. To determine the efficiencies by which the derivatives are metabolized to sialic acids, we synthesized and investigated the corresponding cyclopropane derivatives because cyclopropenes are not stable under the analysis conditions. From these experiments, it became obvious that N-(cycloprop-2-en-1-ylcarbonyl)-modified (Cp-modified) mannosamine has the highest metabolic acceptance. However, carbamate-linked N-(2-methylcycloprop-2-en-1-ylmethyloxycarbonyl)-modified (Cyoc-modified) mannosamine despite its lower metabolic acceptance results in the same cell-surface labeling intensity due to its superior reactivity in the DAinv reaction. Based on the high incorporation efficiency of the Cp derivative we synthesized and investigated two new Cp-modified glucosamine and galactosamine derivatives. Both compounds lead to comparable, distinct cell-surface staining after MGE. We further found that the amide-linked Cp-modified glucosamine derivative but not the Cyoc-modified glucosamine is metabolically converted to the corresponding sialic acid.
February, 8, 2019
Analysis of modification and proteolytic targeting by the ubiquitin-like modifier FAT10
Annette Aichem, Annika N Böhm, Nicola Catone, Gunter Schmidtke and Marcus Groettrup
Methods
8 February 2019, published online
In their in-depth article researchers from Project C01 give detailed methodological information about the handling of the ubiquiti-like modifier FAT10.
Abstract
The ubiquitin-like modifier FAT10 (also called ubiquitin D (UBD)) interacts noncovalently with a substantial number of proteins and also gets covalently conjugated to many substrate proteins, leading to their degradation by the 26S proteasome. FAT10 comprises two loosely folded ubiquitin-like domains that are connected by a flexible linker, and this unusual structure makes it highly prone to aggregation. Here, we report methods to purify high amounts of soluble recombinant FAT10 for various uses, such as in vitro FAT10ylation assays. In addition, we describe how to generate and handle overexpressed as well as endogenous FAT10 in cellulo for use in immunoprecipitations, Western blot analyses, and FAT10 degradation studies.
February, 4, 2019
The ubiquitin-like modifier FAT10 stimulates the activity of the deubiquitylating enzyme OTUB1
Johanna Bialas, Annika N Böhm, Nicola Catone, Annette Aichem and Marcus Groettrup
J Biol Chem
4 February 2019, published online
Scientists for Project C01 demonstrate that the ubiquitin-like modifier FAT10 controls the stbility and the functionality of the deubiquitylase OTUB1 and thus also influences ubiquitylation processes.
Abstract
The deubiquitylation of target proteins is mediated by DUBs such as OTUB1 which plays an important role in the immune response, cell cycle progression and DNA repair. Within these processes OTUB1 reduces the ubiquitylation of target proteins in two distinct ways, either by using its catalytic DUB activity or in a non–catalytic manner by inhibiting the E2 conjugating enzyme. Here, we show that the ubiquitin-like modifier FAT10 is regulating the OTUB1 stability and functionality in different manners. Covalent FAT10ylation of OTUB1 results in its proteasomal degradation whereas a non-covalent interaction stabilizes OTUB1. We provide evidence that OTUB1 directly interacts with FAT10 and the E2 conjugating enzyme USE1. This interaction strongly stimulates the OTUB1 DUB activity towards K48-linked diubiquitin. Furthermore, the non-covalent interaction between FAT10 and OTUB1 not only enhances its isopeptidase activity towards K48-linked ubiquitin moieties but also strengthens its non-catalytic activity in reducing K63 polyubiquitylation of its target protein TRAF3. Additionally, the cellular clearance of overall polyubiquitylation by OTUB1 was strongly stimulated through the presence of FAT10. Addition of FAT10 also led to an increased interaction between OTUB1 and its cognate E2 UbcH5B implying a function of FAT10 in the inhibition of polyubiquitylation. Overall, these data indicate that FAT10 does not only play a role in covalent modification and leading its substrates to proteasomal degradation, but that it also regulates stability and functionality of target proteins by interacting in a non-covalent manner. Thereby FAT10 is able to exert a major influence on ubiquitylation processes.
January, 30, 2019
A tetracycline-dependent ribozyme switch allows conditional induction of gene expression in Caenorhabditis elegans
Lena A Wurmthaler, Monika Sack, Karina Gense, Jörg S Hartig and Martin Gamerdinger
Nat Commun
30 January 2019, published online
In a collaborative effort researchers from CRC 969 Project A05 and Project A07 developed tetracycline-dependent RNA-based genetic switches for use in the eukaryotic model organism Caenorhabditis elegans. These switches allow tissue-specific gene expression and the temporal control of gene expression during all developmental stages of the nematode.
Abstract
The nematode Caenorhabditis elegans represents an important research model. Convenient methods for conditional induction of gene expression in this organism are not available. Here we describe tetracycline-dependent ribozymes as versatile RNA-based genetic switches in C. elegans. Ribozyme insertion into the 3’-UTR converts any gene of interest into a tetracycline-inducible gene allowing temporal and, by using tissue-selective promoters, spatial control of expression in all developmental stages of the worm. Using the ribozyme switches we established inducible C. elegans polyglutamine Huntington’s disease models exhibiting ligand-controlled polyQ-huntingtin expression, inclusion body formation, and toxicity. Our approach circumvents the complicated expression of regulatory proteins. Moreover, only little coding space is necessary and natural promoters can be utilized. With these advantages tetracycline-dependent ribozymes significantly expand the genetic toolbox for C. elegans.
January, 23, 2019
Immunoproteasome inhibition induces plasma cell apoptosis and preserves kidney allografts by activating the unfolded protein response and suppressing plasma cell survival factors.
Jun Li, Julia Körner, Michael Basler, Thomas Brunner, Christopher J Kirk and Marcus Groettrup
Kidney Int
23 January 2019, published online
Researchers from Project C01 together with collaborators from the University of Konstanz, the Biotechnology Institute Thurgau and Kezar Life Sciences report about a new strategy to reduce alloantibody production and allograft rejection by inhibition of the immunoproteasome.
Abstract
Chronic antibody-mediated rejection is the leading cause of allograft dysfunction and loss after kidney transplantation, and current immunosuppressive regimens fail to target the plasma cells that produce alloantibodies. We previously showed that treatment with the immunoproteasome inhibitor ONX 0914 prevented the expansion of plasma cells and prevented chronic allograft nephropathy and organ failure after kidney transplantation in rats, but the mechanism has remained elusive. In the current study, we confirmed a long-term reduction in alloantibody production and improvements in allograft histology in rats treated with ONX 0914 or with the broad-spectrum proteasome inhibitor bortezomib. Plasma cells from allotransplanted rats expressed immunoproteasomes at high levels. Immunoproteasome inhibition with ONX 0914 led to ubiquitin-conjugate accumulation, activation of the unfolded protein response, and induction of apoptosis in plasma cells. In addition, ONX 0914 suppressed the expression of adhesion molecules (VLA-4 and LFA-1), plasma cell survival factors (APRIL and IL-6), and IFN-γ-inducible chemokines in bone marrow, while the APRIL receptor BCMA, the IL-6 receptor, and the chemokine receptors CXCR4 and CXCR3 were down-regulated on plasma cells. Taken together, immunoproteasome inhibition blocked alloantibody production by inducing apoptosis of plasma cells through activating the unfolded protein response and suppressing plasma cell survival factors in the bone marrow.
January, 16, 2019
Expanding the genetic code for site-directed spin-labeling.
Theresa Braun, Malte Drescher and Daniel Summerer
Int J Mol Sci
16 January 2019, published online
Geneticall encoded noncanonical amino acids as a site directed spin label in combination with electron paramagnetic resonance could be used to measure protein structure in living cells. Scientists from Project C03 give an overview about this field of research.
Abstract
Site-directed spin labeling (SDSL) in combination with electron paramagnetic resonance (EPR) spectroscopy enables studies of the structure, dynamics, and interactions of proteins in the noncrystalline state. The scope and analytical value of SDSL–EPR experiments crucially depends on the employed labeling strategy, with key aspects being labeling chemoselectivity and biocompatibility, as well as stability and spectroscopic properties of the resulting label. The use of genetically encoded noncanonical amino acids (ncAA) is an emerging strategy for SDSL that holds great promise for providing excellent chemoselectivity and potential for experiments in complex biological environments such as living cells. We here give a focused overview of recent advancements in this field and discuss their potentials and challenges for advancing SDSL–EPR studies.
January, 12, 2019
Expanding the toolbox of synthetic riboswitches with guanine-dependent aptazymes
Julia Stifel, Maike Spöring and Jörg Hartig
Synth Biol
12 January 2019, published online
Researchers from Project A05 developed new aptazymes that depend on guanine which are operative in both, bacteria and human cells.
Abstract
Artificial riboswitches based on ribozymes serve as versatile tools for ligand-dependent gene expression regulation. Advantages of these so-called aptazymes are their modular architecture and the comparably little coding space they require. A variety of aptamer-ribozyme combinations were constructed in the past 20 years and the resulting aptazymes were applied in diverse contexts in prokaryotic and eukaryotic systems. Most in vivo functional aptazymes are OFF-switches, while ON-switches are more advantageous regarding potential applications in e.g. gene therapy vectors. We developed new ON-switching aptazymes in the model organism Escherichia coli and in mammalian cell culture using the intensely studied guanine-sensing xpt aptamer. Utilizing a high-throughput screening based on fluorescence-activated cell sorting in bacteria we identified up to 9.2-fold ON-switches and OFF-switches with a dynamic range up to 32.7-fold. For constructing ON-switches in HeLa cells, we used a rational design approach based on existing tetracycline-sensitive ON-switches. We discovered that communication modules responding to tetracycline are also functional in the context of guanine aptazymes, demonstrating a high degree of modularity. Here, guanine-responsive ON-switches with a four-fold dynamic range were designed. Summarizing, we introduce a series of novel guanine-dependent ribozyme switches operative in bacteria and human cell culture that significantly broaden the existing toolbox.
January, 9, 2019
Preparation of clathrin-coated vesicles from Arabidopsis thaliana seedlings
Niccolò Mosesso, Tobias Bläske, Marie-Kristin Nagel, Michael Laumann and Erika Isono
Front Plant Sci
9 January 2019, published online
Scientists from Project C08 describe a new method to isolate clathrin-coated vesicles from plant seedlings.
Abstract
Clathrin coated vesicles (CCVs) mediate endocytosis of plasma membrane proteins and deliver their content to the endosomes for either subsequent recycling to the plasma membrane or transport to the vacuole for degradation. CCVs assemble also at the trans-Golgi network (TGN) and is responsible for the transport of proteins to other membranes. Oligomerization of clathrin and recruitment of adaptor protein complexes promote the budding and the release of CCVs. However, many of the details during plant CCV formation are not completely elucidated. The analysis of isolated CCVs is therefore important to better understand the formation of plant CCVs, their cargos and the regulation of clathrin-mediated transport processes. In this article, we describe an optimized method to isolate CCVs from Arabidopsis thaliana seedlings.
November, 30, 2018
Triple orthogonal labeling of glycans by applying photoclick chemistry
Verena Schart, Jessica Hassenrück, Anne-Katrin Späte, Jeremias EGA Dold, Raphael Fahrner and Valentin Wittmann
ChemBioChem
30 November 2018, published online
Using different approaches to label carbohydrates by reporter groups researchers of Project B05 for the first time triply labeled glycans.
Abstract
Bioorthogonal labeling of multiple biomolecules is of current interest in chemical biology. Metabolic glycoengineering (MGE) has been shown to be an appropriate approach to visualizing carbohydrates. Here, we report that the nitrile imine–alkene cycloaddition (photoclick reaction) is a suitable ligation reaction in MGE. Using a mannosamine derivative with an acrylamide reporter group that is efficiently metabolized by cells and that quickly reacts in the photoclick reaction, we labeled sialic acids on the surface of living cells. Screening of several alkenes showed that a previously reported carbamate‐linked methylcyclopropene reporter that is well suited for the inverse‐electron‐demand Diels–Alder (DAinv) reaction has a surprisingly low reactivity in the photoclick reaction. Thus, for the first time, we were able to triply label glycans by a combination of DAinv, photoclick, and copper‐free click chemistry.
October, 29, 2018
Calcineurin promotes APC/C activation at meiotic exit by acting on both XErp1 and Cdc20
Andreas Heim, Thomas Tischer and Thomas U Mayer
EMBO Rep
29 October 2018, published online
Researchers from CRC 969 Project B01 in collaboration with scientists from MRC Cambridge show that the phosphatase Calcineurin participates in the control of meiotic exit upon fertilization by two independent mechanisms.
Abstract
Vertebrate oocytes await fertilization arrested at metaphase of the second meiotic division. Fertilization triggers a transient calcium wave, which induces the activation of the anaphase‐promoting complex/cyclosome (APC/C) and its co‐activator Cdc20 resulting in the destruction of cyclin B and hence meiotic exit. Two calcium‐dependent enzymes are implicated in fertilization‐induced APC/CCdc20 activation: calcium‐/calmodulin‐dependent kinase type II (CaMKII) and calcineurin (CaN). While the role of CaMKII in targeting the APC/C inhibitor XErp1/Emi2 for destruction is well‐established, it remained elusive how CaN affects APC/CCdc20 activation. Here, we discover that CaN contributes to APC/CCdc20 activation in Xenopus laevis oocytes by two independent but interrelated mechanisms. First, it facilitates the degradation of XErp1 by dephosphorylating it at a site that is part of a phosphorylation‐dependent recruiting motif for PP2A‐B′56, which antagonizes inhibitory phosphorylation of XErp1. Second, it dephosphorylates Cdc20 at an inhibitory site, thereby supporting its APC/C‐activating function. Thus, our comprehensive analysis reveals that CaN contributes to timely APC/C activation at fertilization by both negatively regulating the APC/C inhibitory activity of XErp1 and positively regulating the APC/C‐activating function of Cdc20.
October, 29, 2018
Isotopically site-selected dynamics of a three-stranded β-sheet peptide detected with Temperature-Jump IR-spectroscopy
David Scheerer, Heng Chi, Dan McElheny, Timothy A Keiderling and Karin Hauser
J Phys Chem B
29 October 2018, published online
Scientists from CRC 969 Project A02 together with researchers from the University of Chicago show that folding of a model three-stranded β-sheet peptide proceeds in a multi-step manner.
Abstract
Infrared detected temperature jump (T-jump) spectroscopy and site-specific isotopic labeling were applied to study a model three-stranded β-sheet peptide with the goal of individually probing the dynamics of strand and turn structural elements. This peptide had two DPro-Gly (pG) turn sequences to stabilize the two component hairpins, which were labeled with 13C=O on each of the Gly residues to resolve them spectroscopically. Labeling the second turn on the amide preceding the DPro (Xxx-DPro amide) provided an alternate turn label as a control. Placing 13C=O labels on specific in-strand residues gave shifted modes that overlap the Xxx-DPro amide I’ modes. Their impact could be separated from the turn dynamics by a novel difference-transient analysis approach. FTIR spectra were modeled with DFT-computations which showed the local, isotope-selected vibrations were effectively uncoupled from the other amide I modes. Our T-jump dynamics results, combined with NMR structures and equilibrium spectral measurements, showed the first turn to be most stable and best formed with the slowest dynamics, while the second turn and first strand (N-terminus) had similar dynamics, and the third strand (C-terminus) had the fastest dynamics and was the least structured. The relative dynamics of the strands, Xxx-DPro amides and 13C-labeled Gly residues on the turns also qualitatively corresponded to molecular dynamics (MD) simulations of turn and strand fluctuations. MD trajectories indicated the turns to be bistable, with the first turn being Type I’ and the second turn flipping from I’ to II’. The differences in relaxation times for each turn and the separate strands revealed that the folding process of this turn-stabilized β-sheet structure proceeds in a multi-step process.
September, 05, 2018
Structural dynamics of the E6AP/UBE3A-E6-p53 enzyme-substrate complex
Carolin Sailer, Fabian Offensperger, Alexandra Julier, Kai-Michael Kammer, Ryan Walker-Gray, Matthew G. Gold, Martin Scheffner and Florian Stengel
Scientists from CRC 969 Project A06 and Project B02 in collaboration with researchers from the University College London revealed structural and functional dynamics of the E6AP-E6-p53 enzyme-substrate complex .
Nat Commun
05 September 2018, accepted
Abstract
Deregulation of the ubiquitin ligase E6AP is causally linked to the development of human disease, including cervical cancer. In complex with the E6 oncoprotein of human papillomaviruses, E6AP targets the tumor suppressor p53 for degradation, thereby contributing to carcinogenesis. Moreover, E6 acts as a potent activator of E6AP by a yet unknown mechanism. However, structural information explaining how the E6AP-E6-p53 enzyme-substrate complex is assembled, and how E6 stimulates E6AP, is largely missing. Here, we develop and apply different crosslinking mass spectrometry-based approaches to study the E6AP-E6-p53 interplay. We show that binding of E6 induces conformational rearrangements in E6AP, thereby positioning E6 and p53 in the immediate vicinity of the catalytic center of E6AP. Our data provide structural and functional insights into the dynamics of the full-length E6AP-E6-p53 enzyme-substrate complex, demonstrating how E6 can stimulate the ubiquitin ligase activity of E6AP while facilitating ubiquitin transfer from E6AP onto p53.
August, 02, 2018
Fluorescence lifetime sensitive probes for monitoring ATP cleavage
Daniel Hammler, Andreas Marx and Andreas Zumbusch
Scientists from CRC969 Project B08 report on their development of fluorescent ATP analogues which are suited for fluorescence lifetime measurements.
Chem Eur J
02 August 2018, accepted full
Abstract
Adenosine triphosphate (ATP) probes modified with fluorescence dyes that change their fluorescence properties upon cleavage are an interesting tool for monitoring enzymatic ATP turnover. As a readout parameter, fluorescence lifetime is attractive since it is nearly independent of concentration. In our study we synthesised and investigated eleven different ATP analogues, in which the fluorophores were attached to the γ‐phosphate of ATP. All analogues showed distinctly different fluorescence lifetimes compared to the corresponding values of the free fluorophores. Both fluorescence lifetime increases and decreases upon attachment to ATP were observed. In order to shed light on the photophysical processes governing the lifetime changes we performed photoelectron spectroscopy in air (PESA) to determine HOMO energy levels and time‐resolved fluorescence spectroscopy to obtain rate constants. We present evidence that fluorescence quenching in the compounds tested is dynamic and due to photoinduced electron transfer (PET), whereas fluorescence lifetime increases are caused by stacking interactions between chromophore and the nucleobase reducing non‐radiative relaxation. Finally, we demonstrate that enzymatic cleavage of the ATP analogues presented can be followed by continuous monitoring of fluorescence lifetime changes.
July, 31, 2018
The structure of the ubiquitin-like modifier FAT10 reveals an alternative targeting mechanism for proteasomal degradation
Annette Aichem, Samira Anders, Nicola Catone, Philip Rößler, Sophie Stotz, Andrej Berg, Ricarda Schwab, Sophia Scheuermann, Johanna Bialas, Mira C. Schütz-Stoffregen, Gunter Schmidtke, Christine Peter, Marcus Groettrup and Silke Wiesner
Nat Commun
31 July 2018, accepted
Researchers from CRC 969, Project C01 and Project B09, together with collaborators from the Max-Planxk-Institute in Tübingen and the University of Regensburg, reveal the structure of two domains of the ubiquitin-like modifier FAT10 that is intrinsically instable and has a disordered N-terminus.
Abstract
FAT10 is a ubiquitin-like modifier that directly targets proteins for proteasomal degradation. Here, we report the high-resolution structures of the two individual ubiquitin-like domains (UBD) of FAT10 that are joined by a flexible linker. While the UBDs of FAT10 show the typical ubiquitin-fold, their surfaces are entirely different from each other and from ubiquitin explaining their unique binding specificities. Deletion of the linker abrogates FAT10- conjugation while its mutation blocks auto-FAT10ylation of the FAT10-conjugating enzyme USE1 but not bulk conjugate formation. FAT10- but not ubiquitin-mediated degradation is independent of the segregase VCP/p97 in the presence but not the absence of FAT10’s unstructured N-terminal heptapeptide. Stabilization of the FAT10 UBDs strongly decelerates degradation suggesting that the intrinsic instability of FAT10 together with its disordered N-terminus enables the rapid, joint degradation of FAT10 and its substrates without the need for FAT10 de-conjugation and partial substrate unfolding.
April, 24, 2018
Insights into the aggregation mechanism of polyQ proteins with different glutamine repeat lengths
Tetyana Yushchenko, Elke Deuerling and Karin Hauser
Biophys J
28 April 2018, published online
CRC 969 researchers from Projects A01 and A02 analyzed the mechanism of aggregation of PolyQ proteins. Their results show that aggregation varies depending on the length of the PolyQ repeat.
Abstract
Polyglutamine (polyQ) diseases, including Huntington’s disease, result from the aggregation of an abnormally expanded polyQ repeat in the affected protein. The length of the polyQ repeat is essential for the disease’s onset; however, the molecular mechanism of polyQ aggregation is still poorly understood. Controlled conditions and initiation of the aggregation process are prerequisites for the detection of transient intermediate states. We present an attenuated total reflection Fourier-transform infrared spectroscopic approach combined with protein immobilization to study polyQ aggregation dependent on the polyQ length. PolyQ proteins were engineered mimicking the mammalian N-terminus fragment of the Huntingtin protein and containing a polyQ sequence with the number of glutamines below (Q11), close to (Q38), and above (Q56) the disease threshold. A monolayer of the polyQ construct was chemically immobilized on the internal reflection element of the attenuated total reflection cell, and the aggregation was initiated via enzymatic cleavage. Structural changes of the polyQ sequence were monitored by time-resolved infrared difference spectroscopy. We observed faster aggregation kinetics for the longer sequences, and furthermore, we could distinguish β-structured intermediates for the different constructs, allowing us to propose aggregation mechanisms dependent on the repeat length. Q11 forms a β-structured aggregate by intermolecular interaction of stretched monomers, whereas Q38 and Q56 undergo conformational changes to various β-structured intermediates, including intramolecular β-sheets.
April, 12, 2018
Conformational flexibility within the nascent polypeptide-associated complex enables its interactions with structurally diverse client proteins
Esther M Martin, Matthew P Jackson, Martin Gamerdinger, Karina Gense, Theodoros K Karamanos, Julia R Humes, Elke Deuerling, Alison E Ashcroft, and Sheena E Radford (2018)
J Biol Chem
Scientists from CRC 969 Research Project A07 together with researchers from the University of Leeds demonstrate that in order to bind its substrates the ribosome-associated chaperone nascent polypeptide associated complex NAC adopts various compact and expanded conformations.
Abstract
As newly synthesized polypeptides emerge from the ribosome, it is crucial that they fold correctly. To prevent premature aggregation, nascent chains interact with chaperones that facilitate folding or prevent misfolding until protein synthesis is complete. Nascent polypeptide-associated complex (NAC) is a ribosome-associated chaperone important for protein homeostasis. However, how NAC binds its substrates remains unclear. Using native electrospray ionization MS (ESI MS), limited proteolysis, NMR and cross-linking, we analysed the conformational properties of NAC from Caenorhabditis elegans and studied its ability to bind proteins in different conformational states. Our results revealed that NAC adopts an array of compact and expanded conformations and binds weakly to client proteins that are unfolded, folded, or intrinsically disordered, suggestive of broad substrate compatibility. Of note, we found that this weak binding retards aggregation of the intrinsically disordered protein α-synuclein both in vitro and in vivo. These findings provide critical insights into the structure and function of NAC. Specifically, they reveal the ability of NAC to exploit its conformational plasticity to bind a repertoire of substrates having unrelated sequences and structures independently of actively translating ribosomes.
April, 04, 2018
Orientation of lipids in solid supported lipid bilayers studied by polarized ATR-FTIR spectroscopy
Christian Scheibe and Karin Hauser
Biomed Spectrosc Imaging
04 April 2018, published online
In order to analyze protein-membrane interactions researchers from CRC 969 Project A02 revealed that polarized ATR-FTIR spectroscopy of different lipid bilayers is a useful tool to characterize lipid membranes under various environmental conditions.
Abstract
Solid supported lipid bilayers (SSLB) play an important role as biomimetic membranes to study protein-membrane interactions. We investigated the orientation of lipids in SSLBs at different temperatures and over time. Especially the stability of the lipid bilayer and structural changes upon lipid phase transition were analyzed by polarized ATR-FTIR spectroscopy and with SSLBs of different lipid compositions. The integrity of a lipid bilayer consisting of POPC or a 1:1 mixture of POPC and POPG is conserved over a wide temperature range and over several hours. Furthermore, we were able to monitor changes in the orientation of the lipid alkyl chains upon lipid phase transition for DMPC and DSPC. This study shows that the combination of solid supported lipid bilayers and polarized ATR-FTIR spectroscopy is very powerful to characterize lipid membranes under different environmental conditions. The sensitivity of this technique will be exploited in future studies to analyze the effect of protein-membrane interaction on lipid orientation.
April, 04, 2018
A combined approach of surface passivation and specific immobilization to study biomolecules by ATR-FTIR spectroscopy
Annika Krüger, Alexander Bürkle, Aswin Mangerich and Karin Hauser
Biomed Spectrosc Imaging
04 April 2018, published online
Scientists from CRC969 Project A02 report on a strategy to modify solid surfaces in order to apply ATR-FTIR for studying a wide range of biochemical systems and reactions.
Abstract
Attenuated total reflection Fourier-transform infrared (ATR-FTIR) spectroscopy is a surface-sensitive and label-free technique, which is applied to obtain dynamic structural information of biomolecules. The study of proteins by ATR-FTIR spectroscopy can be impeded by their tendency to adsorb to solid surfaces. Furthermore, the adsorption process of proteins is often accompanied with conformational changes, which can interfere with the intended structural analysis. We efficiently modified a silicon ATR crystal surface with polyethylene glycol and thereby create a protein-repellent surface. To achieve a high sensitivity, which enables the study of small conformational changes of biomolecules, we combine surface passivation with specific immobilization. This is accomplished via the biotin-streptavidin interaction, which is one of the strongest known non-covalent protein-ligand interactions. As a proof of concept we present the specific immobilization of DNA. The modified surface is stable against elevated temperatures and 8 M urea and can therefore be used to study a wide range of biochemical systems and reactions. The surface chemistry is simple and performed under mild conditions, which leads to a high applicability of the presented approach.
March, 07, 2018
Org Biomol Chem
07 March 2018, accepted, full
Abstract
January, 05, 2018
Role of aromatic cross-links in structure and dynamics of model three-stranded β-sheet peptides
David Scheerer, Heng Chi, Dan McElheny, Ayesha Samer, Timothy A Keiderling and Karin Hauser
J Phys Chem A
05 January 2018, published online
Scientists from CRC 969 Project A02 together with researchers from the University of Chicago show that aromatic cross-links can affect the structure and the conformational dynamics of model peptides.
Abstract
A series of closely related peptide sequences that form triple-strand structures was designed with a variation of cross-strand aromatic interactions and spectroscopically studied as models for β-sheet formation and stabilities. Structures of the three-strand models were determined with NMR methods and temperature-dependent equilibrium studies performed using circular dichroism and Fourier transform infrared spectroscopies. Our equilibrium data show that the presence of a direct cross-strand aromatic contact in an otherwise folded peptide does not automatically result in an increased thermal stability and can even distort the structure. The effect on the conformational dynamics was studied with infrared-detected temperature-jump relaxation methods and revealed a high sensitivity to the presence and the location of the aromatic cross-links. Aromatic contacts in the three-stranded peptides slow down the dynamics in a site-specific manner, and the impact seems to be related to the distance from the turn. With a Xxx-DPro linkage as a probe with some sensitivity for the turn, small differences were revealed in the relative relaxation of the sheet strands and turn regions. In addition, we analyzed the component hairpins, which showed less uniform dynamics as compared to the parent three-stranded β-sheet peptides.
November, 06, 2017
Highly motif- and organism-dependent effects of naturally occurring hammerhead ribozyme sequences on gene expression
Lena A Wurmthaler, Benedikt Klauser and Jörg S Hartig
RNA Biology
06 November 2017, published online
Researchers from CRC 969 Project A05 revealed that the effects of naturally occurring hammerhead ribozyme on gene expression are highly variable depending on the organism analyzed.
Abstract
Recent bioinformatics studies have demonstrated a wide-spread occurrence of the hammerhead ribozyme (HHR) and similar small endonucleolytic RNA motifs in all domains of life. It is becoming increasingly evident that such ribozyme motifs participate in important genetic processes in diverse organisms. Although the HHR motif has been studied for more than three decades, only little is known about the consequences of ribozyme activity on gene expression. In the present study we analysed eight different naturally occurring HHR sequences in diverse genetic and organismal contexts. We investigated the influence of active ribozymes incorporated into mRNAs in mammalian, yeast and bacterial expression systems. The experiments show an unexpectedly high degree of organism-specific variability of ribozyme-mediated effects on gene expression. The presented findings demonstrate that ribozyme cleavage profoundly affect gene expression. However, the extent of this effect varies and depends strongly on the respective genetic context. The fast-cleaving type 3 HHRs [CChMVd(-) and sLTSV(-)] generally tended to cause the strongest effects on intracellular gene expression. The presented results are important in order to address potential functions of naturally occurring ribozymes in RNA processing and post-transcriptional regulation of gene expression. Additionally, our results are of interest for biotechnology and synthetic biology approaches that aim at the utilisation of self-cleaving ribozymes as widely applicable tools for controlling genetic processes.
October, 17, 2017
Identification of ubiquitin chain interacting proteins
Xiaohui Zhao, Joachim Lutz, Eva Höllmüller, Martin Scheffner, Andreas Marx and Florian Stengel
Angew Chem Int Ed
17 October 2017, accepted
Scientists from CRC969 Project B03 report on their development of an approach to generate all seven homogeneous ubiquitin chains in large quantities.
Abstract
Ubiquitylation, the modification of proteins by ubiquitin (Ub), is one of the most prevalent and versatile post-translational modifications in eukaryotic cells. As Ub also serves as its own substrate, proteins can be modified by numerous different Ub chains, in which the individual moieties are linked via one or several of the seven lysines of Ub. Homogeneous Ub chains, in which the moieties are sequentially linked via the same residue, have been most extensively studied. Yet, due to their restricted availability, the functions of Ub chains linked via K27, K29 or K33 are poorly understood. We have developed an approach that, for the first time, allows the generation of all seven homogeneous Ub chains in large quantities. We show that the chains enable the identification of Ub chain binding proteins by affinity-based proteomics. The potential of our approach is demonstrated by the identification of previously unknown interaction partners of K27-, K29-, and K33-linked Ub chains.
October, 05, 2017
Simultaneous IR-spectroscopic observation of α-synuclein, lipids, and solvent reveals an alternative membrane-induced oligomerization pathway
Mohammad A Fallah, Hanne R Gerding, Christian Scheibe, Malte Drescher, Christiaan Karreman, Stefan Schildknecht, Marcel Leist and Karin Hauser
Chembiochem
05 October 2017, accepted
Using label-free infrared spectroscopy CRC969 researchers from Project A02 and Project C05 discovered that the pathway of aggregation of the protein alpha-synuclein is influenced by the presence of lipid membranes.
Abstract
The intrinsically disordered protein α-synuclein (αS), a known pathogenic factor for Parkinson’s disease, can adopt defined secondary structures when interacting with membranes or during fibrillation. The αS-lipid interaction and the implications of this process for aggregation and damage to membranes are still poorly understood. Therefore, we established a label-free infrared (IR) spectroscopic approach to simultaneously monitor αS conformation and membrane integrity. IR showed its unique sensitivity for identifying distinct α-structured aggregates. A comparative study of wildtype αS and the naturally occurring splicing variant αS Δexon3 yielded new insights into the membrane’s capability of altering aggregation pathways.
September, 11, 2017
Mechanisms for restraining cAMP-dependent protein kinase revealed by subunit quantitation and novel crosslinking approaches
Ryan Walker-Gray, Florian Stengel and Matthew G Gold
Proc Natl Acad Sci USA
11 September 2017, published online
Head of research of CRC969 Project A06 together with colleagues from University College London, UK, analyzed the mechanism by which the activity of protein kinase A is restrained to prevent uncontrolled protein phosphorylation.
Abstract
Protein phosphorylation by cyclic AMP-dependent protein kinase (PKA) underlies key cellular processes, including sympathetic stimulation of heart cells, and potentiation of synaptic strength in neurons. Unrestrained PKA activity is pathological, and an enduring challenge is to understand how the activity of PKA catalytic subunits is directed in cells. We developed a light-activated cross-linking approach to monitor PKA subunit interactions with temporal precision in living cells. This enabled us to refute the recently proposed theory that PKA catalytic subunits remain tethered to regulatory subunits during cAMP elevation. Instead, we have identified other features of PKA signaling for reducing catalytic subunit diffusion and increasing recapture rate. Comprehensive quantitative immunoblotting of protein extracts from human embryonic kidney cells and rat organs reveals that regulatory subunits are always in large molar excess of catalytic subunits (average ∼17-fold). In the majority of organs tested, type II regulatory (RII) subunits were found to be the predominant PKA subunit. We also examined the architecture of PKA complexes containing RII subunits using cross-linking coupled to mass spectrometry. Quantitative comparison of cross-linking within a complex of RIIβ and Cβ, with or without the prototypical anchoring protein AKAP18α, revealed that the dimerization and docking domain of RIIβ is between its second cAMP binding domains. This architecture is compatible with anchored RII subunits directing the myristylated N terminus of catalytic subunits toward the membrane for release and recapture within the plane of the membrane.
July, 04, 2017
Room-temperature in-cell EPR spectroscopy: alpha-Synuclein disease variants remain intrinsically disordered in the cell
Julia Cattani, Vinod Subramaniam and Malte Drescher
Phys Chem Chem Phys
04 July 2017, published online
The conformation of the intrinsically disordered human alpha-Synuclein protein was analyzed in Xenopus laevis oocytes by researchers of CRC969 Project C03 in collaboration with a scientist from the University of Amsterdam by EPR spectroscopy.
Abstract
Human alpha-Synuclein (aS), implicated in Parkinson’s disease, adopts a rich variety of different conformations depending on the macromolecular context. In order to unravel its pathophysiological role, monitoring its intracellular conformational state and identifying differences for the disease variants is crucial. Here, we present an intracellular spectroscopy approach based on a systematic spin-labeling site-scan in combination with intracellular electron paramagnetic resonance spectroscopy determining conformations on a molecular scale. A quantitative and model-based data analysis revealed that the vast majority of aS, be it wild-type or disease variants A30P or A53T, exists in the monomeric intrinsically disordered form in the cell.
April, 20, 2017
One enzyme, three metabolites: Shewanella algae controls siderophore production via the cellular substrate pool
Sina Rütschlin, Sandra Gunesch and Thomas Böttcher
Cell Chem Biol
20 April 2017, published online
The production of a particular siderophore from Shewanelle algae was investigated by researchers of CRC969 Project C06 and unravel a unique evolutionary strategy toward metabolite diversity.
Abstract
Shewanella algae B516 produces avaroferrin, an asymmetric hydroxamate siderophore, which has been shown to inhibit swarming motility of Vibrio alginolyticus. We aimed to elucidate the biosynthesis of this siderophore and to investigate how S. algae coordinates the production of avaroferrin and its two symmetric counterparts. We reconstituted the reaction in vitro with the main enzyme AvbD and the putative biosynthetic precursors, and demonstrate that multispecificity of this enzyme results in the production of all three cyclic hydroxamate siderophores that were previously isolated as natural products from S. algae. Surprisingly, purified AvbD exhibited a clear preference for the larger cadaverine-derived substrate. In live cells, however, siderophore ratios are maximized toward avaroferrin production, and we demonstrate that these siderophore ratios are the result of a regulation on substrate pool level, which may allow rapid evolutionary adaptation to environmental changes. Our results thereby give insights into a unique evolutionary strategy toward metabolite diversity.
March, 29, 2017
Alpha-Synuclein disease mutations are structurally defective and locally affect membrane binding
Martha Robotta, Julia Cattani, Juliana C Martins, Vinod Subramaniam and Malte Drescher
J Am Chem Soc
29 March 2017, published online
Researchers of Project C03 used electron paramagnetic resonance spectroscopy in combination with site-directed spin labeling to investigate the effect of single point mutations of alpha-Synuclein on the characteristics of membrane binding of this protein. Analysis of wild type and variants of the protein associated with familial Parkinson´s disease revealed that the disease-related alterations in alpha-Synuclein result in locally distorted membrane binding properties of the protein (see also here).
Abstract
The intrinsically disordered human protein alpha-Synuclein (αS) has a prominent role in Parkinson’s disease (PD) pathology. Several familial variants of αS are correlated with inherited PD. Disease mutations have been shown to have an impact on lipid membrane binding. Here, using electron paramagnetic resonance spectroscopy in combination with site-directed spin labeling, we show that familial PD-associated variants are structurally defective in membrane binding and alter the local binding properties of the protein.
March, 23, 2017
Biomolecular dynamics studied with IR-spectroscopy using quantum cascade lasers combined with nanosecond perturbation techniques
Alexander Popp, David Scheerer, Benjamin Heck and Karin Hauser
Spectrochim Acta A Mol Biol Spectrosc
23 March 2017, published online
Scientists from Research Project A02 report on their studies on early protein folding events using fast perturbation techniques.
Abstract
Early events of protein folding can be studied with fast perturbation techniques triggering non-equilibrium relaxation dynamics. A nanosecond laser-excited pH-jump or temperature-jump (T-jump) was applied to initiate helix folding or unfolding of poly-l-glutamic acid (PGA). PGA is a homopolypeptide with titratable carboxyl side-chains whose protonation degree determines the PGA conformation. A pH-jump was realized by the photochemical release of protons and induces PGA folding due to protonation of the side-chains. Otherwise, the helical conformation can be unfolded by a T-jump. We operated under conditions where PGA does not aggregate and temperature and pH are the regulatory properties of its conformation. The experiments were performed in such a manner that the folding/unfolding jump proceeded to the same PGA conformation. We quantified the increase/decrease in helicity induced by the pH-/T-jump and demonstrated that the T-jump results in a relatively small change in helical content in contrast to the pH-jump. This is caused by the strong pH-dependence of the PGA conformation. The conformational changes were detected by time-resolved single wavelength IR-spectroscopy using quantum cascade lasers (QCL). We could independently observe the kinetics for α-helix folding and unfolding in PGA by using different perturbation techniques and demonstrate the high sensitivity of time-resolved IR-spectroscopy to study protein folding mechanisms.
March, 20, 2017
Dienophile-modified mannosamine derivatives for metabolic labeling of sialic acids: A comparative study
Jeremias EGA Dold, Jessica Pfotzer, Anne-Katrin Späte and Valentin Wittmann
Chembiochem
20 March 2017, published online
Scientists from Project B05 compare the efficiency of metabolic labeling of sialic acids by different mannosamine derivatives.
Abstract
Sialic acids play an important role in numerous cell adhesion processes and sialylation levels are known to be altered under certain pathogenic conditions such as cancer. Metabolic glycoengineering with mannosamine derivatives is a convenient way to introduce non-natural chemical reporter groups into sialylated glycoconjugates offering the opportunity to label sialic acids using bioorthogonal ligation chemistry. The labeling intensity not only depends on the rate of the ligation reaction but also on the extent to which the natural sialic acids are replaced by the modified ones, i.e. the incorporation efficiency. Here we present a comparative study of eight mannosamine derivatives featuring terminal alkenes as chemical reporter groups that can be labeled by an inverse-electron-demand Diels-Alder (DAinv) reaction. The derivatives differ in chain length as well as the type of linkage (comprising carbamates, amides, and a urea) that connects the terminal alkene to the sugar. As a general trend, increasing chain lengths result in higher DAinv reactivity and at the same time reduced incorporation efficiency. Carbamates are better accepted than amides with the same chain length; nevertheless do the latter result in more intense cell-surface staining visible in life-cell fluorescence microscopy. Finally, a urea derivative was shown to be accepted.
March, 01, 2017
Phosphate-modified nucleotides for monitoring enzyme activity
Susanne Ermert, Andreas Marx and Stephan S Hacker
Top Curr Chem
01 March 2017, published online
Scientists from Research Project B08 report on their synthesis of various types of reporter nucleotides to monitor the activity of different kinds of enzymes.
Abstract
Nucleotides modified at the terminal phosphate position have been proven to be interesting entities to study the activity of a variety of different protein classes. In this chapter, we present various types of modifications that were attached as reporter molecules to the phosphate chain of nucleotides and briefly describe the chemical reactions that are frequently used to synthesize them. Furthermore, we discuss a variety of applications of these molecules. Kinase activity, for instance, was studied by transfer of a phosphate modified with a reporter group to the target proteins. This allows not only studying the activity of kinases, but also identifying their target proteins. Moreover, kinases can also be directly labeled with a reporter at a conserved lysine using acyl-phosphate probes. Another important application for phosphate-modified nucleotides is the study of RNA and DNA polymerases. In this context, single-molecule sequencing is made possible using detection in zero-mode waveguides, nanopores or by a Förster resonance energy transfer (FRET)-based mechanism between the polymerase and a fluorophore-labeled nucleotide. Additionally, fluorogenic nucleotides that utilize an intramolecular interaction between a fluorophore and the nucleobase or an intramolecular FRET effect have been successfully developed to study a variety of different enzymes. Finally, also some novel techniques applying electron paramagnetic resonance (EPR)-based detection of nucleotide cleavage or the detection of the cleavage of fluorophosphates are discussed. Taken together, nucleotides modified at the terminal phosphate position have been applied to study the activity of a large diversity of proteins and are valuable tools to enhance the knowledge of biological systems.
February, 22, 2017
Xenopus laevis Kif18A is a highly processive kinesin required for meiotic spindle integrity
Martin M Möckel, Andreas Heim, Thomas Tischer and Thomas U Mayer
Open Biol
22 February 2017, published online
In this study, researchers from Project B07 report on their characterization of the Kinesin-8 orthologue of human Kif18A, XI_Kif18A, in Xenopus laevis.
Abstract
The assembly and functionality of the mitotic spindle depends on the coordinated activities of microtubule-associated motor-proteins of the dynein and kinesin superfamily. Our current understanding of the function of motor-proteins is significantly shaped by studies using Xenopus laevis egg extract as its open structure allows complex experimental manipulations hardly feasible in other model systems. Yet, the Kinesin-8 orthologue of human Kif18A has not been described in Xenopus laevis so far. Here, we report the cloning and characterization of Xenopus laevis (Xl) Kif18A. Xenopus Kif18A is expressed during oocyte maturation and its depletion from meiotic egg extract results in severe spindle defects. These defects can be rescued by wildtype Kif18A, but not Kif18A lacking motor-activity or the C-terminus. Single molecule microscopy assays revealed that Xl_Kif18A possesses high processivity, which depends on an additional C-terminal microtubule-binding site. Human tissue culture cells depleted of endogenous Kif18A display mitotic defects, which can be rescued by wildtype, but not tail-less Xl_Kif18A. Thus, Xl_Kif18A is the functional orthologue of human Kif18A whose activity is essential for the correct function of meiotic spindles in Xenopus oocytes.
January, 10, 2017
Different enzymatic processing of γ-phosphoramidate and γ-phosphoester-modified ATP-analogues
Susanne Ermert, Stephan M. Hacker, Alexander Buntru, Martin Scheffner, Christof R. Hauck and Andreas Marx
Chembiochem
10 January 2017, published online
Scientists from CRC969 research Projects B08 and B06 analyzed the propensity of processing γ-phosphoester- and γ-phosphoramidate-modified ATP-analogues in ATPases from different enyzme classes.
Abstract
Monitoring the activity of ATP-consuming enzymes provides the basis for elucidating their modes of action and regulation. Although a number of ATP analogues have been developed for this, their scope is restricted because of the limited acceptance by respective enzymes. In order to clarify which kind of phosphate-modified ATP analogues are accepted by the α-β-phosphoanhydride-cleaving ubiquitin-activating enzyme 1 (UBA1) and the β-γ-phosphoanhydride-cleaving focal adhesion kinase (FAK), we tested phosphoramidate- and phosphoester-modified ATP analogues. UBA1 and FAK were able to convert phosphoramidate-modified ATP analogues, even with a bulky modification like biotin. In contrast, a phosphoester-modified analogue was poorly accepted. These results demonstrate that minor variations in the design of ATP analogues for monitoring ATP utilization have a significant impact on enzymatic acceptance.
December, 20, 2016
Newly translated proteins are substrates for ubiquitin, ISG15, and FAT10
Valentina Spinnenhirn, Annegret Bitzer, Annette Aichem and Marcus Groettrup
FEBS Lett
20 December 2016 published online
Researchers from CRC969-project C01show that the ubiquitin-like modifier FAT10 is conjugated to nascent proteins.
Abstract
The ubiquitin-like modifier, FAT10, is involved in proteasomal degradation and antigen processing. As ubiquitin and the ubiquitin-like modifier, ISG15, cotranslationally modify proteins, we investigated whether FAT10 could also be conjugated to newly synthesized proteins. Indeed, we found that nascent proteins are modified with FAT10, but not with the same preference for newly synthesized proteins as observed for ISG15. Our data show that puromycin-labeled polypeptides are strongly modified by ISG15 and less intensely by ubiquitin and FAT10. Nevertheless, conjugates of all three modifiers copurify with ribosomes. Taken together, we show that unlike ISG15, ubiquitin and FAT10 are conjugated to a similar degree to newly translated and pre-existing proteins.
December, 5, 2016
Multivalent contacts of the Hsp70 Ssb contribute to its architecture on ribosomes and nascent chain interaction
Marie A. Hanebuth, Roman Kityk, Sandra J. Fries, Alok Jain, Allison Kriel, Veronique Albanese, Tancred Frickey, Christine Peter, Matthias P. Mayer, Judith Frydman and Elke Deuerling

Scientists from Project A01 (Deuerling group) and Project B09 (Peter group)
Nat Commun
05 December 2016, published online
CRC969-Researchers from Projects A01 and B09 in collaboration with scientists from the University of Stanford solved the long-standing question about the structural basis and functional mode of recruitment of the Hsp70 chaperone Ssb to ribosomes in yeast cells.
Abstract
Hsp70 chaperones assist de novo folding of newly synthesized proteins in all cells. In yeast, the specialized Hsp70 Ssb directly binds to ribosomes. The structural basis and functional mode of recruitment of Ssb to ribosomes is not understood. Here, we present the molecular details underlying ribosome binding of Ssb in Saccharomyces cerevisiae. This interaction is multifaceted, involving the co-chaperone RAC and two specific regions within Ssb characterized by positive charges. The C-terminus of Ssb mediates the key contact and a second attachment point is provided by a KRR-motif in the substrate binding domain. Strikingly, ribosome binding of Ssb is not essential. Autonomous ribosome attachment becomes necessary if RAC is absent, suggesting a dual mode of Ssb recruitment to nascent chains. We propose, that the multilayered ribosomal interaction allows positioning of Ssb in an optimal orientation to the tunnel exit guaranteeing an efficient nascent polypeptide interaction.
September, 29, 2016
Analyzing structure-function relationships of artificial and cancer-associated PARP1 variants by reconstituting TALEN-generated HeLa PARP1 knock-out cells
Lisa Rank, Sebastian Veith, Eva C. Gwosch, Janine Demgenski, Magdalena Ganz, Marjolijn C. Jongmans, Christopher Vogel, Arthur Fischbach, Stefanie Buerger, Jan M.F. Fischer, Tabea Zubel, Anna Stier, Christina Renner, Michael Schmalz, Sascha Beneke, Marcus Groettrup, Roland P. Kuiper, Alexander Bürkle, Elisa Ferrando-May and Aswin Mangerich
Nucleic Acids Res
29 September 2016, published online
CRC 969-Researchers, Project C01 and former Project B04, together with colleagues from Univerity of Leiden and University of Utrecht, report on their studies on deletion of PARP1 by using TALEN-mediated gene targeting in HeLa cells which allows new research on PARylation.
Abstract
Genotoxic stress activates PARP1, resulting in the post-translational modification of proteins with poly(ADP-ribose) (PAR). We genetically deleted PARP1 in one of the most widely used human cell systems, i.e. HeLa cells, via TALEN-mediated gene targeting. After comprehensive characterization of these cells during genotoxic stress, we analyzed structure–function relationships of PARP1 by reconstituting PARP1 KO cells with a series of PARP1 variants. Firstly, we verified that the PARP1\E988K mutant exhibits mono-ADP-ribosylation activity and we demonstrate that the PARP1\L713F mutant is constitutively active in cells. Secondly, both mutants exhibit distinct recruitment kinetics to sites of laser-induced DNA damage, which can potentially be attributed to non-covalent PARP1–PAR interaction via several PAR binding motifs. Thirdly, both mutants had distinct functional consequences in cellular patho-physiology, i.e. PARP1\L713F expression triggered apoptosis, whereas PARP1\E988K reconstitution caused a DNA-damage-induced G2 arrest. Importantly, both effects could be rescued by PARP inhibitor treatment, indicating distinct cellular consequences of constitutive PARylation and mono(ADP-ribosyl)ation. Finally, we demonstrate that the cancer-associated PARP1 SNP variant (V762A) as well as a newly identified inherited PARP1 mutation (F304L\V762A) present in a patient with pediatric colorectal carcinoma exhibit altered biochemical and cellular properties, thereby potentially supporting human carcinogenesis. Together, we establish a novel cellular model for PARylation research, by revealing strong structure–function relationships of natural and artificial PARP1 variants.
July, 28, 2016
Real-time cellular imaging of protein poly(ADP-ribos)ylation
Annette Buntz, Sarah Wallrodt, Eva Gwosch, Michael Schmalz, Sascha Beneke, Elisa Ferrando-May, Andreas Marx and Andreas Zumbusch
Angew Chemie Int Ed
28 July 2016, published online
Researchers from CRC 969 Project B08 and former Project B04 publish a novel technique to monitor poly(ADP-ribos)ylation in living cells.
Abstract
Poly(ADP-ribos)ylation (PARylation) is an important posttranslational protein modification, and is involved in major cellular processes such as gene regulation and DNA repair. Its dysregulation has been linked to several diseases, including cancer. Despite its importance, methods to observe PARylation dynamics within cells are rare. By following a chemical biology approach, we developed a fluorescent NAD+ analogue that proved to be a competitive building block for protein PARylation in vitro and in cells. This allowed us to directly monitor the turnover of PAR in living cells at DNA damage sites after near-infrared (NIR) microirradiation. Additionally, covalent and noncovalent interactions of selected target proteins with PAR chains were visualized in cells by using FLIM-FRET microscopy. Our results open up new opportunities for the study of protein PARylation in real time and in live cells, and will thus contribute to a better understanding of its significance in a cellular context.
July, 11, 2016
Direct monitoring of β-sheet formation in the outer membrane protein TtoA assisted by TtOmp85
Katharina Henke, Wolfram Welte and Karin Hauser
Biochemistry
11 July 2016, published online
CRC 969-researchers, Project A02 and former Project A03, published their studies on the folding mechanism of the outer membrane protein TtoA via ATR-FTIR.
Abstract
Attenuated total reflection Fourier-transform infrared (ATR-FTIR) spectroscopy was applied to investigate the folding of an outer membrane protein, TtoA, assisted by TtOmp85, both from the thermophilic eubacterium Thermus thermophilus. To directly monitor the formation of β-sheet structure in TtoA and to analyze the function of TtOmp85, we immobilized unfolded TtoA on an ATR crystal. Interaction with TtOmp85 initiated TtoA folding as shown by time-dependent spectra recorded during the folding process. Our ATR-FTIR experiments prove that TtOmp85 possesses specific functionality to assist β-sheet formation of TtoA. We demonstrate the potential of this spectroscopic approach to study the interaction of outer membrane proteins in vitro and in a time-resolved manner.
June, 17, 2016
Exploring the potential of Norbornene-modified Mannosamine derivatives for metabolic glycoengineering
Anne-Katrin Späte, Jeremias E.G.A. Dold, Ellen Batroff, Verena F. Schart, Daniel E. Wieland, Oliver R. Baudendistel and Valentin Wittmann
ChemBioChem
17 June 2016, published online
CRC 969-researchers, Project B05, publish studies on the use of norbornenes for metabolic glycoengineering.
Abstract
Metabolic glycoengineering (MGE) allows the introduction of unnaturally modified carbohydrates into cellular glycans and their visualization through bioorthogonal ligation. Alkenes, for example, have been used as reporters that can react through inverse-electron-demand Diels–Alder cycloaddition with tetrazines. Earlier, norbornenes were shown to be suitable dienophiles; however, they had not previously been applied for MGE. We synthesized two norbornene-modified mannosamine derivatives that differ in the stereochemistry at the norbornene (exo/endo linkage). Kinetic investigations revealed that the exo derivative reacts more than twice as rapidly as the endo derivative. Through derivatization with 1,2-diamino-4,5-methylenedioxybenzene (DMB) we confirmed that both derivatives are accepted by cells and incorporated after conversion to a sialic acid. In further MGE experiments the incorporated sugars were ligated to a fluorophore and visualized through confocal fluorescence microscopy and flow cytometry.
June, 02, 2016
Synthetic glycosphingolipids for live-cell labeling
Martin Dauner, Ellen Batroff, Verena Bachmann, Christof R. Hauck and Valentin Wittmann
Bioconjugate Chem
02 June 2016, published online
CRC 969-researchers, Project B05 and Project B06, report on their results on the synthesis of novel probes to label cells in vivo.
Abstract
Glycosphingolipids are an important component of cell membranes that are involved in many biological processes. Fluorescently labeled glycosphingolipids are frequently used to gain insight into their localization. However, the attachment of a fluorophore to the glycan part or—more commonly—to the lipid part of glycosphingolipids is known to alter the biophysical properties and can perturb the biological function of the probe. Presented here is the synthesis of novel glycosphingolipid probes with mono- and disaccharide head groups and ceramide moieties containing fatty acids of varying chain length (C4 to C20). These glycosphingolipids bear an azide or an alkyne group as chemical reporter to which a fluorophore can be attached through a bioorthogonal ligation reaction. The fluorescent tag and any linker connected to it can be chosen in a flexible manner. We demonstrate the suitability of the probes by selective visualization of the plasma membrane of living cells by confocal microscopy techniques. Whereas the derivatives with the shorter fatty acids can be directly applied to HEK 293T cells, the hydrophobic glycosphingolipids with longer fatty acids can be delivered to cells using fusogenic liposomes.
April, 15, 2016
Bioorthogonally functionalized NAD+ analogues for in-cell visualization of poly(ADP-ribose) formation
Sarah Wallrodt, Annette Buntz, Yan Wang, Andreas Zumbusch and Andreas Marx
Angew Chem Int Ed
15 April 2016, published online
CRC 969-researchers, Project B08, report on their synthesis of NAD+ analogues to visualize poly(ADP-ribosyl)ation in vivo.
Abstract
Poly(ADP-ribos)ylation (PARylation) is a major posttranslational modification and signaling event in most eukaryotes. Fundamental processes like DNA repair and transcription are coordinated by this transient polymer and its binding to proteins. ADP-ribosyltransferases (ARTs) build complex ADP-ribose chains from NAD+ onto various acceptor proteins. Molecular studies of PARylation thus remain challenging. Herein, we present the development of bioorthogonally functionalized NAD+ analogues for the imaging of PARylation in vitro and in cells. Our results show that 2-modified NAD+ analogues perform remarkably well and can be applied to the in-cell visualization of PARylation simultaneously in two colors. This tool gives insight into the substrate scope of ARTs and will help to further elucidate the biological role of PARylation by offering fast optical, multichannel read-outs.
March, 01, 2016
Anionic surfactants enhance click reaction-mediated protein conjugation with ubiquitin
Daniel Schneider, Tatjana Schneider, Joos Aschenbrenner, Franziska Mortensen, Martin Scheffner and Andreas Marx
Bioorg Med Chem
01 March 2016, Vol 24, 995 – 1001
CRC 969-researchers, Project B03, report on their modification of Cu(I)-catalyzed alkyne-azide cycloaddition that results in higher yields while retaining functionality of conjugates.
Abstract
The Cu(I)-catalyzed alkyne-azide cycloaddition (CuAAC) has become increasingly important in the conjugation chemistry of biomolecules. For example, it is an efficient and convenient method to generate defined ubiquitin-protein conjugates. Here, we investigate the effect of surfactants on the efficiency of CuAAC for chemical protein ubiquitylation. We found that anionic surfactants enhance conjugate formation by up to 10-fold resulting in high yields even at low (i.e., micromolar) concentrations of the reactants. Notably, the herein investigated conjugates are functional and thus properly folded.
February, 09, 2016
Endocytosis and endosomal trafficking of DNA after gene electrotransfer in vitro.
Christelle Rosazza, Hendrik Deschout, Annette Buntz, Kevin Braeckmans, Marie-Pierre Rols and Andreas Zumbusch
Mol Ther Nucleic Acids
09 February 2016, published online
CRC 969-researchers, Project B08, together with scientists from France and Belgium published their studies on the mechanism of internalization of DNA upon electrotransfer.
Abstract
DNA electrotransfer is a successful technique for gene delivery into cells and represents an attractive alternative to virus-based methods for clinical applications including gene therapy and DNA vaccination. However, little is currently known about the mechanisms governing DNA internalization and its fate inside cells. The objectives of this work were to investigate the role of endocytosis and to quantify the contribution of different routes of cellular trafficking during DNA electrotransfer. To pursue these objectives, we performed flow cytometry and single-particle fluorescence microscopy experiments using inhibitors of endocytosis and endosomal markers. Our results show that ~50% of DNA is internalized by caveolin/raft-mediated endocytosis, 25% by clathrin-mediated endocytosis, and 25% by macropinocytosis. During active transport, DNA is routed through multiple endosomal compartments with, in the hour following electrotransfer, 70% found in Rab5 structures, 50% in Rab11-containing organelles and 30% in Rab9 compartments. Later, 60% of DNA colocalizes with Lamp1 vesicles. Because these molecular markers can overlap while following organelles through several steps of trafficking, the percentages do not sum up to 100%. We conclude that electrotransferred DNA uses the classical endosomal trafficking pathways. Our results are important for a generalized understanding of gene electrotransfer, which is crucial for its safe use in clinics.
January, 21, 2016
Site-specific dynamics of ß-sheet peptides with DPro-Gly turns probed by laser-excited temperature-jump infrared spectroscopy
Alexander Popp, David Scheerer, Heng Chi, Timothy A. Keiderling and Karin Hauser
ChemPhysChem
21 January 2016, published online
CRC 969-scientists, Project A02, in collaboration with researchers from China and the USA, publish their results of their time resolved spectroscopic studies on the cconformational dynamics of ß-sheet peptides.
Abstract
Turn residues as well as side-chain interactions play an important role for the folding of ß-sheets. We investigated the conformational dynamics of a three-stranded ß-sheet peptide (DPDP) and a two-stranded ß-hairpin (WVYY-DP) by time-resolved temperature-jump infrared spectroscopy. Both peptide sequences contain DPro-Gly residues that favor a tight ß-turn. The three-stranded ß-sheet (Ac-VFITSDPGKTYTEVDPGOKILQ-NH2) is stabilized by the turn sequences, whereas the ß-hairpin (SWTVEDPGKYTYK-NH2) folding is assisted both by the turn sequence and by hydrophobic cross-strand interactions. Relaxation times after the T-jump were monitored as a function of temperature and occur on a sub-microsecond time scale, DPDP being faster than WVYY-DP. The Xxx-DPro tertiary amide provides a detectable IR band allowing us to site-specifically probe the dynamics. The relative importance of the turn versus the intra-strand stability in ß-sheet formation is discussed.
January, 12, 2016
Visualization of protein-specific glycosylation inside living cells
Franziska Doll, Annette Bunz, Anne-Katrin Späte, Verena F. Schart, Alexander Timper, Waldemar Schrimpf, Christof R. Hauck, Andreas Zumbusch and Valentin Wittmann
Angew Chemie Int Ed
12 January 2016, published online
CRC 969-scientists, Project B05, in collaboration with projects B06 and B08, report on their analysis of protein glycosylation inside living cells via fluorescence lifetime imaging microscopy.
Abstract
Protein glycosylation is a ubiquitous post-translational modification that is involved in the regulation of many aspects of protein function. In order to uncover the biological roles of this modification, imaging the glycosylation state of specific proteins within living cells would be of fundamental importance. To date, however, this has not been achieved. Herein, we demonstrate protein-specific detection of the glycosylation of the intracellular proteins OGT, Foxo1, p53, and Akt1 in living cells. Our generally applicable approach relies on Diels–Alder chemistry to fluorescently label intracellular carbohydrates through metabolic engineering. The target proteins are tagged with enhanced green fluorescent protein (EGFP). Förster resonance energy transfer (FRET) between the EGFP and the glycan-anchored fluorophore is detected with high contrast even in presence of a large excess of acceptor fluorophores by fluorescence lifetime imaging microscopy (FLIM).
November, 01, 2015
Protein phosphatase 1 is essential for Greatwall inactivation at mitotic exit
Andreas Heim, Anja Konietzny and Thomas u Mayer
EMBO Rep
01 November 2015, Vol 16, 1501 – 1510
CRC 969-scientists, Project B01, provide an answer to the question how the level of phosphorylation of cell cycle regulators is controlled to allow exit from mitosis. The cover of this issue of EMBO Reports shows a graphical abstract of the study which was also commented in a News and Views article by Satoru Mochida (Vol 16, 1411 – 1412).

Abstract
Entry into mitosis is mediated by the phosphorylation of key cell cycle regulators by cyclin‐dependent kinase 1 (Cdk1). In Xenopus embryos, the M‐phase‐promoting activity of Cdk1 is antagonized by protein phosphatase PP2A‐B55. Hence, to ensure robust cell cycle transitions, Cdk1 and PP2A‐B55 must be regulated so that their activities are mutually exclusive. The mechanism underlying PP2A‐B55 inactivation at mitotic entry is well understood: Cdk1‐activated Greatwall (Gwl) kinase phosphorylates Ensa/Arpp19, thereby enabling them to bind to and inhibit PP2A‐B55. However, the re‐activation of PP2A‐B55 during mitotic exit, which is essential for cell cycle progression, is less well understood. Here, we identify protein phosphatase PP1 as an essential component of the PP2A‐B55 re‐activation pathway in Xenopus embryo extracts. PP1 initiates the re‐activation of PP2A‐B55 by dephosphorylating Gwl. We provide evidence that PP1 targets the auto‐phosphorylation site of Gwl, resulting in efficient Gwl inactivation. This step is necessary to facilitate subsequent complete dephosphorylation of Gwl by PP2A‐B55. Thus, by identifying PP1 as the phosphatase initiating Gwl inactivation, our study provides the molecular explanation for how Cdk1 inactivation is coupled to PP2A‐B55 re‐activation at mitotic exit.
September, 24, 2015
Click chemistry for targeted protein ubiquitylation and ubiquitin chain formation
Daniel Rösner, Tatjana Schneider, Daniel Schneider, Martin Scheffner and Andreas Marx
Nat Protocols
Availably online 24 September 2015 | Print edition: Vol 10, 1594-1611
CRC 969-scientists, Project B03, report on a convenient method using click chemistry to generate ubiquitin-protein conjugates site-specifically.
Abstract
Herein we describe a simple protocol for the efficient generation of site-specific ubiquitin-protein conjugates using click chemistry. By using two different methods to expand the genetic code, the two bio-orthogonal functionalities that are necessary for CuI-catalyzed azide-alkyne cycloaddition (CuAAC), an alkyne and an azide, are co-translationally incorporated into the proteins of interest with unnatural amino acids. Protein ubiquitylation is subsequently carried out with the purified proteins in vitro by CuAAC. In addition, we provide a protocol for the incorporation of two unnatural amino acids into a single ubiquitin, resulting in a ‘bifunctional’ protein that contains both an alkyne and an azide functionality, thereby enabling assembly of free ubiquitin chains as well as ubiquitin chains conjugated to a target protein. Our procedure enables the synthesis of nonhydrolyzable ubiquitin-protein conjugates within 1 week (given that the relevant cDNAs are at hand), and it yields conjugates in milligram quantities from 1-liter expression cultures. The approach described herein is faster and less laborious than other methods, and it requires only standard molecular biology equipment. Moreover, the protocol can be readily adapted to achieve conjugation at any site of any target protein, which facilitates the generation of custom-tailored ubiquitin-protein conjugates.
July, 27, 2015
Role of ubiquitin and the HPV E6 oncoprotein in E6AP-mediated protein ubiquitination
Franziska Mortensen, Daniel Schneider, Tanja Barbic, Anna Sladewska-Marquardt, Simone Kühnle, Andreas Marx and Martin Scheffner
Proc Natl Acad Sci USA
Availably online 27 July 2015
CRC 969-scientists, Project B03, report on their studies on the control of the ubiquitin ligase E6 associated protein (E6AP) by noncovalent interactions with ubiquitin and allosteric activators such as the HPV E6 oncoprotein.
Abstract
Deregulation of the ubiquitin ligase E6 associated protein (E6AP) encoded by the UBE3A gene has been associated with three different clinical pictures. Hijacking of E6AP by the E6 oncoprotein of distinct human papillomaviruses (HPV) contributes to the development of cervical cancer, whereas loss of E6AP expression or function is the cause of Angelman syndrome, a neurodevelopmental disorder, and increased expression of E6AP has been involved in autism spectrum disorders. Although these observations indicate that the activity of E6AP has to be tightly controlled, only little is known about how E6AP is regulated at the posttranslational level. Here, we provide evidence that the hydrophobic patch of ubiquitin comprising Leu-8 and Ile-44 is important for E6AP-mediated ubiquitination, whereas it does not affect the catalytic properties of the isolated catalytic HECT domain of E6AP. Furthermore, we show that the HPV E6 oncoprotein rescues the disability of full-length E6AP to use a respective hydrophobic patch mutant of ubiquitin for ubiquitination and that it stimulates E6AP-mediated ubiquitination of Ring1B, a known substrate of E6AP, in vitro and in cells. Based on these data, we propose that E6AP exists in at least two different states, an active and a less active or latent one, and that the activity of E6AP is controlled by noncovalent interactions with ubiquitin and allosteric activators such as the HPV E6 oncoprotein.
May, 15, 2015
Mad2 inhibitor-1 (M2I-1): A small molecule protein-protein interaction inhibitor targeting the mitotic spindle assembly checkpoint
Johanna Kastl, Joachim Braun, Andreas Prestel, Heiko M Möller, Thomas Huhn and Thomas u Mayer
ACS Chem Biol
Accepted 15 May 2015
CRC 969969-scientists, Project B01, report on their identification of a small molecule inhibitor that targets an essential component of the mitotic spindle assembly checkpoint.
Abstract
The genetic integrity of each organism depends on the faithful segregation of its genome during mitosis. To meet this challenge, a cellular surveillance mechanism, termed the spindle assembly checkpoint (SAC), evolved that monitors the correct attachment of chromosomes and blocks progression through mitosis if corrections are needed. While the central role of the SAC for genome integrity is well established, its functional dissection has been hampered by the limited availability of appropriate small molecule inhibitors. Using a fluorescence polarization-based screen, we identify Mad2 inhibitor-1 (M2I-1), the first small molecule inhibitor targeting the binding of Mad2 to Cdc20, an essential protein-protein interaction (PPI) within the SAC. Based on computational and biochemical analyses, we propose that M2I-1 disturbs conformational dynamics of Mad2 critical for complex formation with Cdc20. Cellular studies revealed that M2I-1 weakens the SAC response, indicating that the compound might be active in cells. Thus, our study identifies the SAC specific complex formation between Mad2 and Cdc20 as a protein-protein interaction that can be targeted by small molecules.
April, 12, 2015
Not4-dependent translation repression is important for cellular protein homeostasis in yeast
Steffen Preissler, Julia Reuther, Miriam Koch, Annika Scior, Michael Bruderek, Tancred Frickey and Elke Deuerling
EMBO J
Accepted 12 April 2015
CRC 969-scientists, Project A01, report on their finding that the protein Not4 which is part of the multifunctional Ccr4-Not complex in yeast contributes to negative regulation of protein synthesis.
Abstract
Translation of aberrant or problematic mRNAs can cause ribosome stalling which leads to production of truncated or defective proteins. Therefore, cells evolved cotranslational quality control mechanisms that eliminate these transcripts and target arrested nascent polypeptides for proteasomal degradation. Here we show that Not4, which is part of the multifunctional Ccr4-Not complex in yeast, associates with polysomes and contributes to negative regulation of protein synthesis. Not4 is involved in translation repression of transcripts that cause transient ribosome stalling. The absence of Not4 affected global translation repression upon nutrient withdrawal, enhanced the expression of arrested nascent polypeptides and caused constitutive protein folding stress and aggregation. Similar defects were observed in cells with impaired mRNA-decapping protein function and in cells lacking the mRNA decapping activator and translation repressor Dhh1. The results suggest a role for Not4 together with components of the decapping machinery in regulation of protein expression on the mRNA level and emphasize the importance of translation repression for maintenance of proteome integrity.
April, 10, 2015
The principle of antagonism ensures protein targeting specificity at the endoplasmic reticulum
Martin Gamerdinger, Marie Anne Hanebuth, Tancred Frickey and Elke Deuerling
Science
10 April 2015, Vol 348:201-207
Researchers of the CRC 969, Project A01, have revealed what is necessary to prevent erroneous protein transport. Two competing activities ensure that proteins safely arrive at their intended destination – in cell organelles like the mitochondria and the endoplasmic reticulum (ER), in particular. The team succeeded in uncovering that, in contrast to the prevailing view, successful protein transport requires not only the signal recognition particle (SRP), but also the nascent polypeptide-associated complex (NAC).
Abstract
The sorting of proteins to the appropriate compartment is one of the most fundamental cellular processes. We found that in the model organism Caenorhabditis elegans, correct cotranslational endoplasmic reticulum (ER) transport required the suppressor activity of the nascent polypeptide-associated complex (NAC). NAC did not affect the accurate targeting of ribosomes to ER translocons mediated by the signal recognition particle (SRP) pathway but inhibited additional unspecific contacts between ribosomes and translocons by blocking their autonomous binding affinity. NAC depletion shortened the life span of Caenorhabditis elegans, caused global mistargeting of translating ribosomes to the ER, and provoked incorrect import of mitochondrial proteins into the ER lumen, resulting in a strong impairment of protein homeostasis in both compartments. Thus, the antagonistic targeting activity of NAC is important in vivo to preserve the robustness and specificity of cellular protein-sorting routes.
March, 13, 2015
Conjugation of the ubiquitin activating enzyme UBE1 with the ubiquitin-like modifier FAT10 targets it for proteasomal degradation
Johanna Bialas, Marcus Groettrup & Annette Aichem
PLoS One
Researchers of the CRC 969, Project C01, report on their studies on the regulatory role of the ubiquitin-like modifier FAT10 in the ubiquitin conjugation pathway.
Abstract
The ubiquitin-like modifier HLA-F adjacent transcript 10 (FAT10) directly targets its substrates for proteasomal degradation by becoming covalently attached via its C-terminal diglycine motif to internal lysine residues of its substrate proteins. The conjugation machinery consists of the bispecific E1 activating enzyme Ubiquitin-like modifier activating enzyme 6 (UBA6), the likewise bispecific E2 conjugating enzyme UBA6-specific E2 enzyme 1 (USE1), and possibly E3 ligases. By mass spectrometry analysis the ubiquitin E1 activating enzyme ubiquitin-activating enzyme 1 (UBE1) was identified as putative substrate of FAT10. Here, we confirm that UBE1 and FAT10 form a stable non-reducible conjugate under overexpression as well as under endogenous conditions after induction of endogenous FAT10 expression with proinflammatory cytokines. FAT10ylation of UBE1 depends on the diglycine motif of FAT10. By specifically downregulating FAT10, UBA6 or USE1 with siRNAs, we show that UBE1 modification depends on the FAT10 conjugation pathway. Furthermore, we confirm that UBE1 does not act as a second E1 activating enzyme for FAT10 but that FAT10ylation of UBE1 leads to its proteasomal degradation, implying a putative regulatory role of FAT10 in the ubiquitin conjugation pathway.
February, 25, 2015
Fluorescence-based monitoring of ribosome assembly landscapes
Rainer Nikolay, Renate Schloemer, Silke Mueller & Elke Deuerling
BMC Molecular Biology 16, 3
Researchers of the CRC 969, Project A01, report on their strategy to monitor ribosome assembly by differently labeling the ribosomal subunits.
Abstract
Background
Ribosomes and functional complexes of them have been analyzed at the atomic level. Far less is known about the dynamic assembly and degradation events that define the half-life of ribosomes and guarantee their quality control.
Results
We developed a system that allows visualization of intact ribosomal subunits and assembly intermediates (i.e. assembly landscapes) by convenient fluorescence-based analysis. To this end, we labeled the early assembly ribosomal proteins L1 and S15 with the fluorescent proteins mAzami green and mCherry, respectively, using chromosomal gene insertion. The reporter strain harbors fluorescently labeled ribosomal subunits that operate wild type-like, as shown by biochemical and growth assays. Using genetic and chemical perturbations by depleting genes encoding the ribosomal proteins L3 and S17, respectively, or using ribosome-targeting antibiotics, we provoked ribosomal subunit assembly defects. These defects were readily identified by fluorometric analysis after sucrose density centrifugation in unprecedented resolution.
Conclusion
This strategy is useful to monitor and characterize subunit specific assembly defects caused by ribosome-targeting drugs that are currently used and to characterize new molecules that affect ribosome assembly and thereby constitute new classes of antibacterial agents.
December, 23, 2014
pH-jump induced leucine zipper folding beyond the diffusion limit
Mateusz L Donten, Shabir Hassan, Alexander Popp, Jonathan Halter, Karin Hauser & Peter Hamm
The Journal of Physical Chemistry B
Published Online 23 December 2014 | Print Edition: Vol 119, 1425-1432
Researchers of the CRC 969, Project A02, in collaboration with researchers from the University of Zurich, report on their analysis of the folding of a pH-sensitive leucine zipper peptide.
Abstract
The folding of a pH-sensitive leucine zipper, that is, a GCN4 mutant containing eight glutamic acid residues, has been investigated. A pH-jump induced by a caged proton (o-nitrobenzaldehyde, oNBA) is employed to initiate the process, and time-resolved IR spectroscopy of the amide I band is used to probe it. The experiment has been carefully designed to minimize the buffer capacity of the sample solution so that a large pH jump can be achieved, leading to a transition from a completely unfolded to a completely folded state with a single laser shot. In order to eliminate the otherwise rate-limiting diffusion-controlled step of the association of two peptides, they have been covalently linked. The results for the folding kinetics of the cross-linked peptide are compared with those of an unlinked peptide, which reveals a detailed picture of the folding mechanism. That is, folding occurs in two steps, one on an ∼1–2 μs time scale leading to a partially folded α-helix even in the monomeric case and a second one leading to the final coiled-coil structure on distinctively different time scales of ∼30 μs for the cross-linked peptide and ∼200 μs for the unlinked peptide. By varying the initial pH, it is found that the folding mechanism is consistent with a thermodynamic two-state model, despite the fact that a transient intermediate is observed in the kinetic experiment.
November, 17, 2014
Synthesis and biological evaluation of optimized inhibitors of the mitotic kinesin Kif18A
Tobias Strittmatter,
ACS Chemical Biology
Published Online 17 November 2014 | Print edition: Vol 10:554-560
Researchers of the CRC 969, Projects B07 and B08, report on the synthesis of inhibitors of Kif18A that potently inhibit the ATPase activity of the mitotic kinesin.
The mitotic spindle, a highly dynamic structure composed of microtubules, mediates the segregation of the previously duplicated genome into the two nascent daughter cells. Errors in this process contribute to pathology including tumor formation. Key for the shape and function of the mitotic spindle are kinesins, molecular motor proteins that convert chemical energy into mechanical work. Due to their fast mode of action, small molecules are valuable tools to dissect the dynamic functions of kinesins during mitosis. In this study, we report the identification of optimized small molecule inhibitors of the mitotic kinesin Kif18A. Using BTB-1, the first identified Kif18A inhibitor, as lead compound we synthesized a collection of derivatives. We demonstrate that some of the synthesized derivatives potently inhibited the ATPase activity of Kif18A with a half maximal inhibitory concentration (IC50) value in the low micromolar range. In vitro analysis of a panel of Kif18A-related kinesins revealed that the two most potent compounds show improved selectivity compared to BTB-1. Structure-activity relationship studies identified substituents mediating undesired inhibitory effects on microtubule polymerization. In summary, our study provides key insights into the mechanism of action of BTB-1 and its analogs which will have a great impact on the further development of highly selective and bioactive Kif18A inhibitors. Since Kif18A is frequently overexpressed in solid tumors, such compounds are not only of great interest for basic research but have also the potential to open up new strategies for the treatment of human diseases.
November, 13, 2014
Effect of hydrophobic interactions on the folding mechanism of β-hairpins
The Journal of Physical Chemistry
Published Online 13 November 2014 | Print edition: Vol 118:14234-14242
Researchers of the CRC 969, Project A02, report on their investigations on the influence of hydrophobic interactions on the folding pathway and kinetics of protein folding.
Hydrophobic interactions are essential in stabilizing protein structures. How they affect the folding pathway and kinetics, however, is less clear. We used time-resolved infrared spectroscopy to study the dynamics of hydrophobic interactions of β-hairpin variants of the sequence Trpzip2 (SWTWENGKWTWK-NH2) that is stabilized by two cross-strand Trp-Trp pairs. The hydrophobicity strength was varied by substituting the tryptophans pairwise by either tyrosines or valines. Relaxation dynamics were induced by a laser-excited temperature-jump which separately probed for the loss of the cross-strand β-hairpin interaction and the rise of the disordered structure. All substitutions tested result in reduced thermal stability, lower transition temperatures and faster dynamics compared to Trpzip2. However, the changes in folding dynamics depend on the amino acid substituted for Trp. The aromatic substitution of Tyr for Trp results in the same kinetics for the unfolding of sheet and growth of disorder, with similar activation energies, independent of the substitution position. Substitution of Trp with a solely hydrophobic Val results in even faster kinetics than substitution with Tyr, but is additionally site-dependent. If the hairpin has a Val pair close to its termini, the rate constants for loss of sheet and gain of disorder are the same, but if the pair is close to the turn, the sheet and disorder components show different relaxation kinetics. The Trp→Val substitutions reveal that hydrophobic interactions alone weakly stabilize the hairpin structure, but adding edge-to-face aromatic interaction strengthens it, and both modify the complex folding process.
October, 8, 2014
Terminal alkenes as versatile chemical reporter groups for metabolic oligosaccharide engineering
Sophie Schöllkopf,
Chemistry European Journal
Published Online 8 October 2014 | Print edition: Vol 20:16502-16508
Researchers of the CRC 969, Project B05, report on their studies on metabolic oligosaccharide engineering using terminal alkenes.
The Diels–Alder reaction with inverse electron demand (DAinv reaction) of 1,2,4,5-tetrazines with electron rich or strained alkenes was proven to be a bioorthogonal ligation reaction that proceeds fast and with high yields. An important application of the DAinv reaction is metabolic oligosaccharide engineering (MOE) which allows the visualization of glycoconjugates in living cells. In this approach, a sugar derivative bearing a chemical reporter group is metabolically incorporated into cellular glycoconjugates and subsequently derivatized with a probe by means of a bioorthogonal ligation reaction. Here, we investigated a series of new mannosamine and glucosamine derivatives with carbamate-linked side chains of varying length terminated by alkene groups and their suitability for labeling cell-surface glycans. Kinetic investigations showed that the reactivity of the alkenes in DAinv reactions increases with growing chain length. When applied to MOE, one of the compounds, peracetylated N-butenyloxycarbonylmannosamine, was especially well suited for labeling cell-surface glycans. Obviously, the length of its side chain represents the optimal balance between incorporation efficiency and speed of the labeling reaction. Sialidase treatment of the cells before the bioorthogonal labeling reaction showed that this sugar derivative is attached to the glycans in form of the corresponding sialic acid derivative and not epimerized to another hexosamine derivative to a considerable extent.
September, 25, 2014
Efficient labelling of enzymatically synthesized vinyl-modified DNA by an inverse-electron-demand Diels-Alder reaction
Chemical Communications
Published Online 22 July 2014 | Print edition: Vol 50:10827-10829
Researchers of the CRC 969, Projects B05 and B08, report on their development of a method to label modified nucleotides efficiently.
Many applications in biotechnology and molecular biology rely on modified nucleotides. Here, we present an approach for the postsynthetic labelling of enzymatically synthesized vinyl-modified DNA by Diels–Alder reaction with inverse electron demand using a tetrazine. Labelling proceeds very efficiently and supersedes several known approaches.
September, 22, 2014
Expanding the scope of cyclopropene reporters for the detection of metabolically engineered glycoproteins by Diels–Alder reactions
Julia Häfner,
Beilstein Journal of Organic Chemistry
Published Online 22 September 2014 | Print edition: Vol 10:2235-2242
Researchers of the CRC 969, Projects B05 and B07, report on the use of cyclopyrene tags to visualize mucin-type O-glycoproteins and O-GlcNAcylated proteins.
Monitoring glycoconjugates has been tremendously facilitated by the development of metabolic oligosaccharide engineering. Recently, the inverse-electron-demand Diels–Alder reaction between methylcyclopropene tags and tetrazines has become a popular ligation reaction due to the small size and high reactivity of cyclopropene tags. Attaching the cyclopropene tag to mannosamine via a carbamate linkage has made the reaction even more efficient. Here, we expand the application of cyclopropene tags to N-acylgalactosamine and N-acylglucosamine derivatives enabling the visualization of mucin-type O-glycoproteins and O-GlcNAcylated proteins through Diels–Alder chemistry. Whereas the previously reported cyclopropene-labeled N-acylmannosamine derivative leads to significantly higher fluorescence staining of cell-surface glycoconjugates, the glucosamine derivative gave higher labeling efficiency with protein preparations containing also intracellular proteins.
September, 10, 2014
Alpha-Synuclein binds to the inner membrane of mitochondria in an α-helical conformation
Marta Robotta, Hanne R. Gerding, Antonia Vogel, Karin Hauser, Stefan Schildknecht, Christiaan Karreman, Marcel Leist, Vinod Subramaniam & Malte Drescher
Chembiochem
Published Online 10 September 2014 |
Researchers of the CRC 969, Projects A01, C03 and C04, report on their studies on the conformation of alpha-synuclein bound to isolated mitochondria.
The human alpha-Synuclein (αS) protein is of significant interest because of its association with Parkinson’s disease and related neurodegenerative disorders. The intrinsically disordered protein (140 amino acids) is characterized by the absence of a well-defined structure in solution. It displays remarkable conformational flexibility upon macromolecular interactions, and can associate with mitochondrial membranes. Site-directed spin-labeling in combination with electron paramagnetic resonance spectroscopy enabled us to study the local binding properties of αS on artificial membranes (mimicking the inner and outer mitochondrial membranes), and to evaluate the importance of cardiolipin in this interaction. With pulsed, twofrequency, double-electron electron paramagnetic resonance (DEER) approaches, we examined, to the best of our knowledge for the first time, the conformation of αS bound to isolated mitochondria.
September, 4, 2014
Dissecting ubiquitin signaling with linkage-defined and protease resistant ubiquitin chains
Tatjana Schneider, Daniel Schneider, Daniel Rösner, Saurav Malhotra, Franziska Mortensen, Thomas U. Mayer, Martin Scheffner & Andreas Marx
Angewandte Chemie International Edition English
Published Online 4 September 2014 |
Researchers of the CRC 969, Projects B03 and B07, report on the development of a new method to generate linkage-defined ubiquitin chains to analyze ubiquitin signaling.
Ubiquitylation is a complex posttranslational protein modification and deregulation of this pathway has been associated with different human disorders. Ubiquitylation comes in different flavors: Besides mono-ubiquitylation, ubiquitin chains of various topologies are formed on substrate proteins. The fate of ubiquitylated proteins is determined by the linkage-type of the attached ubiquitin chains, however, the underlying mechanism is poorly characterized. Herein, we describe a new method based on codon expansion and click-chemistry-based polymerization to generate linkage-defined ubiquitin chains that are resistant to ubiquitin-specific proteases and adopt native-like functions. The potential of these artificial chains for analyzing ubiquitin signaling is demonstrated by linkage-specific effects on cell-cycle progression.
September, 4, 2014
The human oncoprotein and chromatin architectural factor DEK counteracts DNA replication stress
Anja Deutzmann, Magdalena Ganz, Felix Schönenberger, Jörg Vervoorts, Ferdinand Kappes & Elisa Ferrando-May
Oncogene
Accepted 4 September 2014 | Published Online 27 October 2014
Researchers of the CRC 969, Project B04, in collaboration with scientists from the University of Aachen report on their studies on the role of the chromatin architectual factor DEK in the management of DNA replication stress.
DNA replication stress is a major source of DNA strand breaks and genomic instability, and a hallmark of precancerous lesions. In these hyperproliferative tissues, activation of the DNA damage response results in apoptosis or senescence preventing or delaying their development to full malignancy. In cells, in which this antitumor barrier is disabled by mutations (for example, in p53), viability and further uncontrolled proliferation depend on factors that help to cope with replication-associated DNA damage. Replication problems preferentially arise in chromatin regions harboring complex DNA structures. DEK is a unique chromatin architectural factor which binds to non-B-form DNA structures, such as cruciform DNA or four-way junctions. It regulates DNA topology and chromatin organization, and is essential for the maintenance of heterochromatin integrity. Since its isolation as part of an oncogenic fusion in a subtype of AML, DEK has been consistently associated with tumor progression and chemoresistance. How DEK promotes cancer, however, is poorly understood. Here we show that DEK facilitates cellular proliferation under conditions of DNA replication stress by promoting replication fork progression. DEK also protects from the transmission of DNA damage to the daughter cell generation. We propose that DEK counteracts replication stress and ensures proliferative advantage by resolving problematic DNA and/or chromatin structures at the replication fork.
July, 22, 2014
Pre-anaphase chromosome oscillations are regulated by the antagonistic activities of Cdk1 and PP1 on Kif18A
Julia Häfner, Monika I Mayr, Martin M Möckel & Thomas U Mayer
Nature Communications
Published Online 22 July 2014 |
Researchers of the CRC 969, Project B07, report on their identification of a network of regulatory proteins that controls verterbrate chromosome oscillations essential for the fidelity of sister chromatid segregation.
Upon congression at the spindle equator, vertebrate chromosomes display oscillatory movements which typically decline as cells progress towards anaphase. Kinesin-8 Kif18A has been identified as a suppressor of chromosome movements, but how its activity is temporally regulated to dampen chromosome oscillations before anaphase onset remained mysterious. Here, we identify a regulatory network composed of cyclin-dependent kinase-1 (Cdk1) and protein phosphatase-1 (PP1) that antagonistically regulate Kif18A. Cdk1-mediated inhibitory phosphorylation of Kif18A promotes chromosome oscillations in early metaphase. PP1 induces metaphase plate thinning by directly dephosphorylating Kif18A. Chromosome attachment induces Cdk1 inactivation and kinetochore recruitment of PP1a/g. Thus, we propose that chromosome biorientation mediates the alignment of chromosomes at the metaphase plate by tipping the balance in favour of dephosphorylated Kif18A capable of suppressing the oscillatory movements of chromosomes. Notably, interfering with chromosome oscillations severely impairs the fidelity of sister chromatid segregation demonstrating the importance of timely controlled chromosome dynamics for the maintenance of genome integrity.
July, 05, 2014
Poly(ADP-ribose)-mediated interplay of XPA and PARP1 leads to reciprocal regulation of protein function
Jan MF Fischer, Oliver Popp, Daniel Gebhard, Sebastian Veith, Arthur Fischbach, Sascha Beneke, Alfred Leitenstorfer, Jörg Bergemann, Martin Scheffner, Elisa Ferrando-May, Aswin Mangerich & Alexander Bürkle
FEBS Journal
Published Online 05 July 2014 | Print edition 281(16):6325-6341
Researchers of the CRC 969, Projects B02 and B04, report on their studies to analyze the functional significance of non-covalent, high affinity binding of the nucleotide excision repair protein XPA to Poly-ADP-ribose.
Poly(ADP-ribose) (PAR) is a complex and reversible posttranslational modification controlling protein function and localization through covalent modification of or non-covalent binding to target proteins. Previously, we and others characterized the non-covalent, high-affinity binding of the key nucleotide excision repair (NER) protein XPA to PAR. Here, we address the functional relevance of this interaction. First, we confirm that pharmacological inhibition of cellular poly(ADP-ribosyl)ation (PARylation) impairs NER efficacy. Second, we demonstrate that the XPA-PAR interaction is mediated by specific basic amino acids within a highly conserved PAR binding motif, which is overlapping with XPA’s DDB2 and TFIIH interaction domains. Third, biochemical studies reveal a mutual regulation of PARP1 and XPA functions showing that on the one hand, XPA-PAR interaction lowers XPA’s DNA binding affinity, while on the other hand, XPA itself strongly stimulates PARP1 enzymatic activity. Fourth, radiation experiments in U2OS cells demonstrate that PARP inhibition alters the recruitment properties of XPA-GFP to sites of laser-induced DNA damage. In conclusion, our results reveal that XPA and PARP1 regulate each other in a reciprocal and PAR-dependent manner, potentially acting as a fine-tuning mechanism for the spatio-temporal regulation of the two factors during NER.
April, 29, 2014
Thiamine pyrophosphate stimulates acetone activation by Desulfococcus biacutus as monitored by a fluorogenic ATP analogue
ACS Chemical Biology
Published Online 29 April 2014 | Print Edition 9(6):1263-1266
Researchers of the CRC 969, Project B08, together with researchers from the University of Konstanz report on theuse of fluorigenic ATP analogues to analyze bacterial acetone degradation.
Acetone can be degraded by aerobic and anaerobic microorganisms. Studies with the strictly anaerobic sulfate-reducing bacterium Desulfococcus biacutus indicate that acetone degradation by these bacteria starts with an ATP-dependent carbonylation reaction leading to acetoacetaldehyde as the first reaction product. The reaction represents the second example of a carbonylation reaction in the biochemistry of strictly anaerobic bacteria, but the exact mechanism and dependence on cofactors are still unclear. Here, we use a novel fluorogenic ATP analogue to investigate its mechanism. We find that thiamine pyrophosphate is a cofactor of this ATP-dependent reaction. The products of ATP cleavage are AMP and pyrophosphate, providing first insights into the reaction mechanism by indicating that the reaction proceeds without intermediate formation of acetone enol phosphate.
February, 28, 2014
Stability of the Thermus thermophilus outer membrane protein TtoA against heat and denaturants
Katharina Henke, Meike Odermatt, Wolfram Welte & Karin Hauser
Biomedical Spectroscopy and Imaging
Published Online 28 February 2014 | Print edition: Vol 3(1):51-56
Researchers of the CRC 969, Projects A02 and A03, report on their FT-IR spectroscopy and SDS PAGE analysis of the stability of the outer membrane protein TtoA.
TtoA is a major outer membrane protein (Omp) with β-barrel structure from the thermophilic eubacterium T. thermophilus. FT-IR spectroscopy and SDS PAGE analysis were used to monitor the stability of detergent-solubilised TtoA under denaturing conditions. Heat as well as the common denaturants urea and SDS were applied to affect the TtoA structure. The protein has proven to be extremely thermostable in its native form. Denaturants likewise only have little effects on the protein’s structure. In detail, a SDS concentration of 1% as is typical for Laemmli buffer does not have an impact on the structure of TtoA, even in combination with a temperature of 99°C. The same holds true for urea concentrations below 8 M. Only the combination of highly concentrated urea or SDS in combination with incubating the protein at 99°C for 10 minutes leads to a change of the secondary structure in TtoA.
December, 11, 2013
Rapid Labeling of Metabolically Engineered Cell-Surface Glycoconjugates with a Carbamate-Linked Cyclopropene Reporter
Anne-Kathrin Späte, Holger Bußkamp, Andrea Niederwieser, Verena F. Schart, Andreas Marx & Valentin Wittmann
Bioconjugate Chemistry
Published Online 11 December 2013 | Print edition: Vol 25(1):147–154
Researchers of the CRC 969, Projects B05 and B08, report on the synthesis of a novel N-acyl-mannosamine derivative that cells incorporate into glycoconjugates. This derivative undergoes rapid inverse-electron-demand Diels-Alder reactions with labeling reagents.
Metabolic oligosaccharide engineering is a valuable tool to monitor cellular carbohydrates. Here, we report the synthesis of a novel N-acyl-mannosamine derivative bearing a methylcyclopropene tag that is attached to the sugar via a carbamate moiety. This derivative undergoes rapid Diels-Alder reaction with inverse electron demand. We demonstrate that the cell’s biosynthetic machinery incorporates this non-natural mannosamine derivative into glycoconjugates that can, subsequently, be labeled within less than 10 min with a new sulfo-Cy3–tetrazine-conjugate. Using this tetrazine dye conjugate for the detection of the methylcyclopropene-tagged mannosamine derivative we could achieve dual labeling of two different metabolically incorporated sugars in which a Diels-Alder reaction with inverse electron demand and a strain-promoted azide-alkyne cycloaddition are carried out simultaneously in a single step.
November, 26, 2013
Length-dependent Conformational Transitions of Polyglutamine Repeats as Molecular Origin of Fibril Initiation
Benjamin Heck, Franziska Doll & Karin Hauser
Biophysical Chemistry
Published Online 26 November 2013 | Print edition: Vol 185:47–57
Researchers of the CRC 969, Project A02, report on their spectroscopic analyses of the conformation of PolyQ model peptides.
Polyglutamine (polyQ) sequences are found in a variety of proteins with normal function. However, their repeat expansion is associated with a number of neurodegenerative diseases, also called polyQ diseases. The length of the polyQ sequence, varying in the number of consecutive glutamines among different diseases, is critical for inducing fibril formation. We performed a systematic spectroscopic study to analyze the conformation of polyQ model peptides in dependence of the glutamine sequence lengths (K2QnK2 with n=10, 20, 30). Complementary FTIR- and CD-spectra were measured in a wide concentration range and repeated heating and cooling cycles revealed the thermal stability of formed β-sheets. The shortest glutamine sequence K2Q10K2 shows solely random structure for concentrations up to 10mg/ml. By increasing the peptide length to K2Q20K2, a significant fraction of β-sheet is observed even at low concentrations of 0.01mg/ml. The higher the concentration, the more the structural composition is dominated by the intermolecular β-sheet. The formation of highly thermostable β-sheet is much more pronounced in K2Q30K2. K2Q30K2 precipitates at a concentration of 0.3mg/ml. Our spectroscopic study shows that the aggregation tendency is enhanced with increased glutamine repeat expansion and that the concentration plays another critical factor in the β-sheet formation.
October, 31, 2013
Fluorogenic ATP Analogues for Online Monitoring of ATP Consumption: Observing Ubiquitin Activation in Real-Time
Stephan M. Hacker, Dana Pagliarini, Thomas Tischer, Norman Hardt, Daniel Schneider, Martin Mex, Thomas U. Mayer, Martin Scheffner & Andreas Marx
Angewandte Chemie, International Edition
Published Online 17 September 2013 | Print edition: Vol 52(45):11916 –11919
Researchers of the CRC 969, Projects B08 and B02, report on the development of new ATP-based probe.
The authors present a conceptually new method to monitor the activity of the ATP-consuming enzyme UBA1. The method is based on fluorogenic ATP analogues undergoing a large change in fluorescence characteristics upon enzymatic processing. The method allows monitoring and quantifying activation of ubiquitin and ubiquitin-like proteins by their cognate E1s in the absence of the downstream enzyme cascade in real-time in a facile and parallel fashion. Additionally, the presented approach is applicable to study effects of protein interactions on activity as exemplarily shown for E1-E2-interactions.
May, 13, 2013
Role of the ubiquitin ligase E6AP/UBE3A in controlling levels of the synaptic protein Arc
Simone Kühnle, Benedikt Mothes, Konstantin Matentzoglu & Martin Scheffner
Proceedings of the National Academy of Sciences USA
Published Online 13 May 2013 | Print edition: Vol 110(22):8888-8893
Researchers of the CRC 969, Project B02, report on their studies on the role of E6AP/UBE3A in the control of Arc protein levels.
Abstract
Inactivation of the ubiquitin ligase E6 associated protein (E6AP) encoded by the UBE3A gene has been associated with development of the Angelman syndrome. Recently, it was reported that in mice, loss of E6AP expression results in increased levels of the synaptic protein Arc and a concomitant impaired synaptic function, providing an explanation for some phenotypic features of Angelman syndrome patients. Accordingly, E6AP has been shown to negatively regulate activity-regulated cytoskeleton-associated protein (Arc) and it has been suggested that E6AP targets Arc for ubiquitination and degradation. In our study, we provide evidence that Arc is not a direct substrate for E6AP and binds only weakly to E6AP, if at all. Furthermore, we show that down-regulation of E6AP expression stimulates estradiol-induced transcription of the Arc gene. Thus, we propose that Arc protein levels are controlled by E6AP at the transcriptional rather than at the posttranslational level.
April, 23, 2013
Ribozyme-based transfer RNA switches for post-transcriptional control of amino acid identity in protein synthesis
Athanasios Saragliadis & Jörg S. Hartig
Journal of the American Chemical Society
Published Online 23 April 2013 | Print edition: Vol 135(22):8222-8226
Researchers of the CRC 969, Project A05, report on their novel strategy to generate individual and distinct protein variants by using switchable transfer RNAs.
Abstract
Protein mutants are studied in a variety of contexts in the life sciences. However, individual mutations need to be generated in order to transcribe and translate the respective protein variants. Here, we introduce a novel strategy for controlling the incorporation of different amino acids in response to an amber stop codon by utilizing switchable designer transfer RNAs in Escherichia coli.
April, 19, 2013
The nascent polypeptide-associated complex is a key regulator of proteostasis
Janine Kirstein-Miles, Annika Scior, Elke Deuerling & Rick Morimoto
EMBO J
Accepted 18 March 2013 | Published Online 19 April 2013
Researchers of the CRC 969, Project A01, together with colleagues from the Rice Institute for Biomedical Research in Evanston report on the localization of the nascent polypeptide associated complex under different stress conditions and reveal its role as a cellular proteostasis sensor.
Abstract
The adaptation of protein synthesis to environmental and physiological challenges is essential for cell viability. Here, we show that translation is tightly linked to the protein-folding environment of the cell through the functional properties of the ribosome bound chaperone NAC (nascent polypeptide-associated complex). Under non-stress conditions, NAC associates with ribosomes to promote translation and protein folding. When proteostasis is imbalanced, NAC relocalizes from a ribosome-associated state to protein aggregates in its role as a chaperone. This results in a functional depletion of NAC from the ribosome that diminishes translational capacity and the flux of nascent proteins. Depletion of NAC from polysomes and re-localisation to protein aggregates is observed during ageing, in response to heat shock and upon expression of the highly aggregation-prone polyglutamine-expansion proteins and Aβ-peptide. These results demonstrate that NAC has a central role as a proteostasis sensor to provide the cell with a regulatory feedback mechanism in which translational activity is also controlled by the folding state of the cellular proteome and the cellular response to stress.
April, 03, 2013
Improving bioorthogonal protein ubiquitylation by click reaction
Daniel Schneider, Tatjana Schneider, Daniel Rösner, Martin Scheffner & Andreas Marx
Bioorg Med Chem
Published Online 3 April 2013
Researchers of the CRC 969, Project B03, report on a technique to generate high amounts of site-specifically mono-ubiquitylated proteins.
Abstract
Posttranslational modification of proteins with ubiquitin (ubiquitylation) regulates numerous cellular processes. Besides functioning as a signal for proteasomal degradation, ubiquitylation has also non-proteolytic functions by altering the biochemical properties of the modified protein. To investigate the effect(s) of ubiquitylation on the properties of a protein, sufficient amounts of homogenously and well-defined ubiquitylated proteins are required. Here, we report on the elaboration of a method for the generation of high amounts of site-specifically mono-ubiquitylated proteins. Firstly, a one-step affinity purification scheme was developed for ubiquitin containing the unnatural amino acid azidohomoalanine at the C-terminal position. This ubiquitin was conjugated in a click reaction to recombinant DNA polymerase β, equipped with an alkyne function at a distinct position. Secondly, addition of defined amounts of SDS to the reaction significantly improved product formation. With these two technical improvements, we have developed a straight forward procedure for the efficient generation of site-specifically ubiquitylated proteins that can be used to study the effect(s) of ubiquitylation on the activities/properties of a protein.
March, 06, 2013
Two-Color Glycan Labeling of Live Cells by a Combination of Diels–Alder and Click Chemistry
Andrea Niederwieser, Anne-Katrin Späte, Long Duc Nguyen, Christian Jüngst, Werner Reutter & Valentin Wittmann
Angewandte Chemie, International Edition
Published Online 6 March 2013 | Print edition 8 April 2013: Vol 52(15):4265-4268
Researchers of the CRC 969, Project B05, together with colleagues from the Charité in Berlin-Dahlem report on the labeling of different glycan structures and their simultaneous visualization in live cells.
Abstract
One is not enough: Terminal alkenes are used as chemical reporters and ligation partners for 1,2,4,5-tetrazines in a Diels–Alder reaction with inverse electron demand (DARinv). Combination with strain-promoted azide–alkyne cycloaddition (SPAAC) allows the visualization of two different glycan structures in one experiment.
March, 05, 2013
Fingerprinting differential active site constraints of ATPases
Stephan M. Hacker, Norman Hardt, Alexander Buntru, Dana Pagliarini, Martin Möckel, Thomas U. Mayer, Martin Scheffner, Christof R. Hauck & Andreas Marx
Chemical Science
Received 5 November 2012 | Published online 15 January 2013
Researchers of the CRC 969, Projects B06, B07 and B08, report on their synthesis of ATP probes and the application of these probes to analyze the active site constraints of different ATPases.
Abstract:
The free energy provided by adenosine triphosphate (ATP) hydrolysis is central to many cellular processes and, therefore, the number of enzymes utilizing ATP as a substrate is almost innumerable. Modified analogues of ATP are a valuable means to understand the biological function of ATPases. Although these enzymes have evolved towards binding to ATP, large differences in active site architectures were found. In order to systematically access the specific active site constraints of different ATPases suitable tools are required.
Here, we present the synthesis of six new ATP-based ATPase probes modified at three different positions of the nucleobase and the ribose, respectively. Subsequently, we studied the ATPases focal adhesion kinase FAK, the ubiquitin-activating protein UBA1 and the kinesin Eg5 as examples for ATPases that process ATP by different mechanisms.
We find that for each of these enzymes at least one position in ATP can be modified without loss of acceptance by the enzyme. However, the positions at which modifications are tolerated significantly differ between the studied enzymes allowing fingerprints to be drawn for reactivity. The introduced ATP analogues may form the basis for the design of tailored probes with increased affinity and specificity for a specific ATPase of interest.
October, 22, 2012
Synthesis and Stability of Phosphate Modified ATP Analogues
Stephan M. Hacker, Martin Mex & Andreas Marx
The Journal of Organic Chemistry
Received 7 Sept 2012 | Published online 22 Oct 2012
Researchers of the CRC 969, Project B08, report on their synthesis of phosphate modified ATP analogues.
Abstract:
Nucleotides modified at the phosphate have numerous applications. Nevertheless, the number of attachment modes is limited and little is known about their stability.
Here, we present results on the elaboration of the synthesis of five classes of ATP analogues and studies concerning their stability. We show that the nitrogen-linked ATP analogue is less stable, whereas the oxygen- and novel carbon-linked adenosine tri- and tetraphosphate analogues are stable from pH 3 to 12 rendering them interesting for further applications and designs.
September, 27, 2012
The APC/C Inhibitor XErp1/Emi2 Is Essential for Xenopus Early Embryonic Divisions
Thomas Tischer, Eva Hörmanseder & Thomas U. Mayer
Science
Received 3 Aug 2012 | Accepted 4 Sept 2012 | Published online 27 Sept 2012
Researchers of the CRC 969, Project B01, have elucidated the mechanism of early embryonic cell division control.
Abstract:
Mitotic divisions result from the oscillating activity of cyclin-dependent kinase 1 (Cdk1). Cdk1 activity is terminated by the anaphase-promoting complex/cyclosome (APC/C), a ubiquitin ligase that targets cyclin B for destruction.
In somatic divisions, the early mitotic inhibitor 1 (Emi1) and the spindle assembly checkpoint (SAC) regulate cell cycle progression by inhibiting the APC/C. Early embryonic divisions lack these APC/C-inhibitory components, which raises the question of how those cycles are controlled.
We found that the APC/C-inhibitory activity of XErp1 (also known as Emi2) was essential for early divisions in Xenopus embryos. Loss of XErp1 resulted in untimely destruction of APC/C substrates and embryonic lethality. XErp1’s APC/C-inhibitory function was negatively regulated by Cdk1 and positively by protein phosphatase 2A (PP2A).
Thus, Cdk1 and PP2A operate at the core of early mitotic cell cycles by antagonistically controlling XErp1 activity, which results in oscillating APC/C activity.
April, 12, 2012
Locally resolved membrane binding affinity of the N-terminus of α-Synuclein.
Marta Robotta, Christian Hintze, Stefan Schildknecht, Niels Zijlstra, Christian Jüngst, Christiaan Karreman, Martina Huber, Marcel Leist, Vinod Subramaniam & Malte Drescher
Biochemistry
Received 19 March 2012 | Accepted 12 April 2012 | Published online 12 April 2012
Researchers of the CRC 969, Projects C03 and C04, have analyzed the membrane binding affinity of α-Synuclein by EPR spectroscopy.
Abstract:
α-Synuclein is abundantly present in Lewy bodies, characteristic of Parkinson’s disease. Its exact physiological role has yet to be determined, but mitochondrial membrane binding is suspected to be a key aspect of its function.
Electron paramagnetic resonance spectroscopy in combination with site-directed spin labeling allowed for a locally resolved analysis of the protein-membrane binding affinity for artificial phospho-lipid membranes, supported by a study of binding to isolated mitochondria. The data reveal that the binding affinity of the N-terminus is nonuniform.
March, 26, 2012
FAT10 and NUB1L bind to the VWA domain of Rpn10 and Rpn1 to enable proteasome-mediated proteolysis
Neha Rani, Annette Aichem, Gunter Schmidtke, Stefan G. Kreft & Marcus Groettrup
Nature Communications
Received 16 Nov 2011 | Accepted 16 Feb 2012 | Published 20 Mar 2012
The 18 kD protein FAT10 (HLA-F locus adjacent transcript 10) targets proteins for degradation by the 26S proteasome. It functions similarly to poly-ubiqitin, yet acts ubiquitin-independently. Researchers of the CRC 969, Project C01, have now shown how FAT10 docks to the proteasome to initiate proteolysis.
FAT10 binds to the Rpn10 subunit of the proteasome by interaction of its C-terminal UBL (ubiquitin like) domain with Rpn10´s VWA domain. Upon binding both, FAT10 and its conjugated protein are slowly degraded by the proteasome.
Degradation is substantially accelerated by the scaffold protein NUB1L (NEDD8 ultimate buster-1 long). NUB1L binds to the proteasome at two sites: Its UBL domain interacts with Rpn10, unexpectedly also via Rpn10´s VWA domain which seems to be a novel UBL-binding domain. In addition, NUB1L binds to Rpn1.
The researchers provide a model that explains the accelerating action of NUB1L on degradation of FAT10 and its conjugated protein. It involves the newly discovered binding of NUB1L to Rpn1 to either transfer FAT10 and its conjugated substrates to Rpn10 or to facilitate their rapid degradation.